Cryogenic electron microscopy – beyond the resolution revolution
Issue: Future Tech
09 August 2016 article

Cryo-electron microscopy (cryoEM) has recently emerged as a structural biology technique to rival X-ray crystallography. Technological developments in both instrumentation and software have allowed researchers to determine the structures of macromolecular assemblies at close to atomic resolution. Moreover, assemblies that would be intractable by X-ray methods owing to heterogeneity or disordered regions have become viable targets for structure determination, prompting the wide uptake of a method that, until comparatively recently, was sometimes considered ‘niche’. So, what does the future hold for structural electron microscopy and what does it offer the microbiologist?
The long history of a rapidly emerging method in structural biology
Since its invention in the 1930s, the transmission electron microscope (TEM) has been an invaluable tool to investigate the shapes of macromolecular assemblies and cellular ultrastructure. Although the TEM has long been capable of imaging materials at the atomic scale, specimen preparation requirements limited the achievable resolution in biology. Classical biological transmission electron microscopy preserves cells and tissue by chemical fixation and dehydration, followed by resin embedding and ultra-thin sectioning. Purified macromolecules are likewise imaged in a dehydrated state, in negative stain. Drying, fixation and staining severely compromise the structural integrity of biological assemblies, and high-resolution features are lost.
In the 1980s, researchers working in the laboratory of Jacques Dubochet, at the University of Lausanne, pioneered methods for the preparation of biological material in a frozen hydrated state, by freezing suspensions of macromolecules so rapidly that there is no time for ice crystals to form – a process known as vitrification. This led to a landmark publication describing cryogenic TEM imaging of viruses (Nature paper in Further reading).
Images recorded in the TEM are two-dimensional (2D) projections of the three-dimensional (3D) object. For structure analysis, images of identical objects viewed in different orientations may be computationally processed to determine a 3D density map (3D reconstruction – Fig. 1). Methods to recover 3D structure data from negative-stain TEM images were first developed by David DeRosier and Aaron Klug in the late 1960s, working at the Medical Research Council Laboratory of Molecular Biology in Cambridge. In the late 1980s, the application of computational 3D reconstruction methods to cryoEM images led to the publication of the first low-resolution structures for unstained icosahedral viruses and the birth of a new discipline in the field of structural biology.
FIG. 1. CRYOMICROSCOPY OF A CALICIVIRUS GAVE 2D PROJECTION VIEWS OF RANDOMLY ORIENTED PARTICLE (BACKGROUND). THESE WERE PROCESSED TO PRODUCE A 3D DENSITY MAP (BOTTOM-RIGHT – CENTRAL SLICE THROUGH THE 3D MAP), WHICH CAN BE REPRESENTED AS A COLOURED 3D SURFACE.
From blob-ology to atoms
These early reconstructions provided insights into the quaternary arrangements of larger assemblies and allowed the visualisation of domains in protein structures. Technological innovations in the intervening years led to a steady increase in attainable resolution. Improvements in microscope design and the development of algorithms to correct an imaging artefact known as the contrast-transfer function allowed researchers to determine structures at better than 10 Å (1 nm) resolution in the late 1990s. For the first time this produced reconstructions of sufficient detail to permit the modelling of protein folds under certain optimal conditions. At resolutions around 8–9 Å, α-helices are seen as tubes of density. At 5 Å, β-strands start to resolve and the larger amino acid side-chains become visible.
A step change in the capacity of cryoEM to solve protein structures came about as a consequence of several technological developments over the past five years. First and foremost, the development of second-generation digital cameras for TEM that directly detect electrons as they pass through the sensor. Direct detection devices (DDD) record high-resolution information far more efficiently than the previous CCD-based cameras or photographic film. DDDs also operate at very high frame rates, between 20 and 400 fps. Thus, rather than recording a single frame, micrographs are recorded as movies of many tens of images. At the highest frame rates, DDD cameras can count individual electrons passing through the camera, greatly reducing noise.
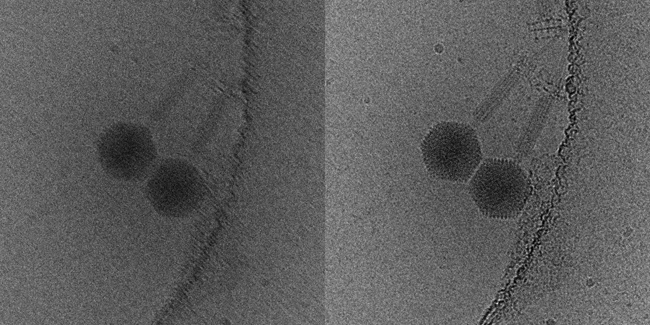
DDDs have not only vastly improved the quality of images recorded in the TEM, they have also informed our understanding of specimen behaviour under the electron beam. Specimen drift had been a major problem in cryoEM, which would severely degrade image quality. Images recorded as movies can be aligned and averaged to compensate for specimen drift during the exposure (Fig. 2). DDD movies have, however, revealed that specimen movement during imaging is much more complex than previously thought. In addition to lateral drift of the specimen, particles also rotate and move relative to each other as the ice layer ‘domes’ under the electron beam, an effect that can also now be corrected in the reconstruction process.
FIG. 2. SPECIMEN DRIFT CAN LEAD TO A BLURRED IMAGE THAT IS UNSUITABLE FOR STRUCTURE ANALYSIS (LEFT). THE HIGH FRAME RATE OF DDD CAMERAS MEANS THAT THIS ARTEFACT CAN BE COMPUTATIONALLY CORRECTED (RIGHT), YIELDING SHARP IMAGES SUITABLE FOR 3D RECONSTRUCTION.
Recent developments in electron microscope design, including more stable lenses and automation, have improved data quality and throughput. Finally, modern software algorithms allow researchers to perform in silico purification: classification of heterogeneous datasets to solve multiple structures or exclude damaged particles from the 3D reconstruction. Taken together, these technological advances have extended attainable resolution to 2–4 Å, a point at which protein models can be built with a high degree of confidence.
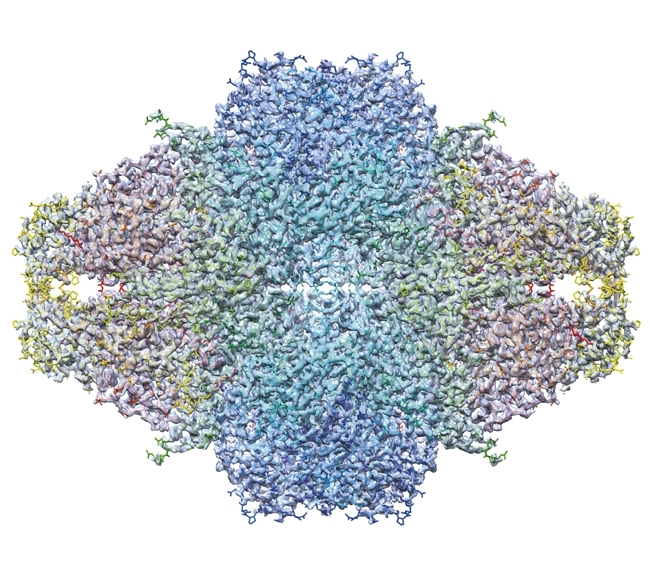
The first de novo structure deposited in the protein data bank (PDB) derived from images recorded in the cryoEM was solved by analysis of 2D crystals of bacteriorhodopsin as early as 1990. It was nearly 20 years before comparable resolutions were achieved for particulate assemblies – helical and icosahedral objects were solved in 2007 and 2008, respectively. The lower symmetry 20S proteasome (a 14-mer) and asymmetric ribosome were solved in 2013 using DDD image data. More recently, astonishing resolutions have been achieved such as the 2.2 Å structure of β-galactosidase (Fig. 3). The improved contrast of DDD cameras operated in electron counting modes means that smaller assemblies are also becoming viable targets for analysis, as illustrated by the recent structure of the ~170 kDa human γ-secretase.
FIG. 3. THE 2.2 Å STRUCTURE OF Β-GALACTOSIDASE.
Technological advances in cryoEM have led to a seismic shift in the field of structural biology, bringing about a rapid expansion in the number of close to atomic-resolution structures in the EM data bank (Fig. 4). Such structures can be determined swiftly, often only requiring one or two days of microscope time followed by several weeks of computation. This is illustrated by the speed with which structures of Zika virus have been produced (Fig. 5).
FIG. 4. A PLOT TO SHOW THE NUMBER OF STRUCTURES DEPOSITED IN THE EM DATABANK EACH YEAR THAT ARE BETTER THAN 10 (BLUE) AND 4 (ORANGE) Å RESOLUTION. THIS ILLUSTRATES THE RECENT EXPLOSION OF HIGH-RESOLUTION STRUCTURES DETERMINED BY CRYOEM.
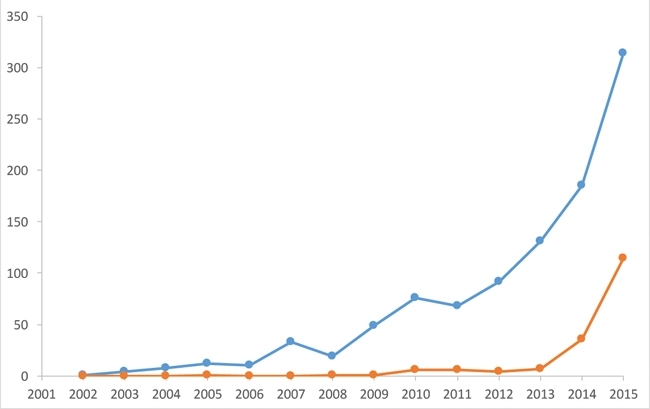
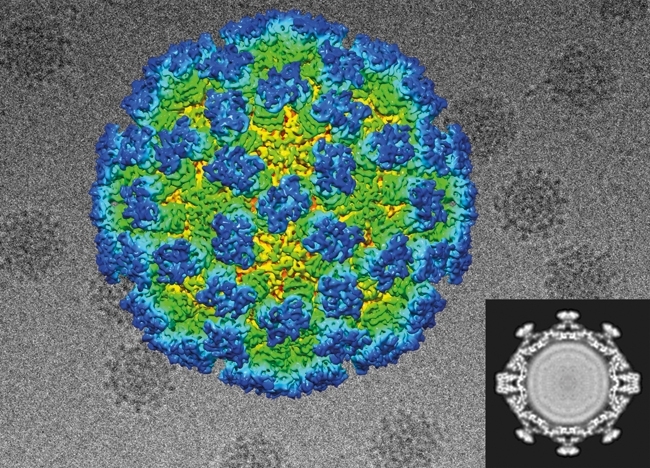
FIG. 5. THE 3.8 Å STRUCTURE OF ZIKA VIRUS.
Taking structural biology into the cell
Electron microscopy as a tool in structural biology might be said to have come of age in recent years, finally realising its potential as a high-resolution method. In my view, the cryomicroscopy revolution has just begun. Single particle reconstruction still has considerable potential for improvement; minimising specimen movement should lead to improved resolution. Larger, faster DDD cameras with improved imaging performance will increase throughput and data quality, while software methods to deal with domain movement within assemblies are beginning to allow researchers to characterise dynamic processes.
The TEM is a versatile instrument, however. In addition to high-resolution analysis of purified assemblies, cryo-electron tomography (cryoET) allows us to investigate the structure of unique entities. This is achieved by rotating the specimen in the electron beam and recording an image every 1–3 degrees. The resulting tilt-series of images is then processed to compute a 3D reconstruction or tomogram. Repeated features within the tomogram may be extracted, brought into common register and averaged – subtomogram averaging.
As well as providing insights into the structure of pleomorphic structures such as enveloped viruses, cryoET raises the tantalising prospect of in situ structure determination: the capacity to solve the structure of an object functioning in the cellular environment. Assemblies such as flagellar motors or bacteriophages can be investigated in situ through tomography of frozen-hydrated intact bacteria, albeit presently at lower resolution. In situ structure analysis of macromolecular assemblies within eukaryotic cells is also becoming a possibility. In virology, many aspects of the replication cycle, for example viral attachment, entry, trafficking through cellular organelles and morphogenesis, are becoming open to investigation. This remains challenging, as the size of objects that may be imaged in the TEM is limited by the penetrating power of the electron beam. Several approaches have been taken to overcome this; firstly, it is possible to propagate viruses in cells that have thin regions and may be imaged directly (Fig. 6). Many of the most interesting processes occur at the heart of the cell and in large organelles. This requires the cell to be sectioned in a frozen-hydrated state. While this is far from trivial, tantalising first steps are being taken to achieve this.
FIG. 6. A CENTRAL SLICE THROUGH A TOMOGRAM OF A NEURONAL CELL THAT IS INFECTED WITH HERPES SIMPLEX VIRUS TYPE 1, CAPSIDS (BLACK ARROWS) ARE SEEN TRAVELLING ALONG MICROTUBULES (WHITE ARROWS).

CryoEM has opened new horizons in structural biology research that will have broad application to address fundamental questions in microbiology. Rapid calculation of high-resolution structures for purified macromolecular assemblies, as well as the developing capacity to determine structures in situ, are tremendously powerful additions to the microbiologist’s toolbox that will increasingly inform our understanding of molecular structure and function in the coming years.
DAVID BHELLA
Centre for Virus Research, Sir Michael Stoker Building, 464 Bearsden Road, Glasgow G61 1QH, UK
[email protected]
@DavidBhella
FURTHER READING
Adrian, M. & others (1984). Cryo-electron microscopy of viruses. Nature 308, 32–36.
Bartesaghi, A. & others (2015). 2.2 Å resolution cryo-EM structure of β-galactosidase in complex with a cell-permeant inhibitor. Science 348, 1147–1151.
Sirohi, D. & others (2016). The 3.8 Å resolution cryo-EM structure of Zika virus. Science 352, 467–470.
Image: Fig. 1. Michaela Conley and David Bhella, University of Glasgow. Fig. 2. Diana Alves and Toby Jenkins, University of Bath; David Bhella, University of Glasgow. Fig. 3. Science 348, 1147–1151 (2015), EMDB 2984 and PDB 5a1a. Fig. 5. Science 352, 467–470 (2016), EMDB 8116. Fig. 6. Rebecca Lauder and Frazier Rixon, University of Glasgow..