Putting Oxford Nanopore Technology to the Test
Hello, I’m Taylor Schababerle, a master’s student in the Diagnostic Genetics and Genomics program at the University of Texas MD Anderson Cancer Center School of Health Professions.
Reproducibility in science has been a central topic for nearly as long as scientific inquiry has existed. It is not enough for a single researcher to obtain a compelling result, findings only become robust when they can be independently replicated and validated. Scientists, by nature, tend to be skeptical of results that don’t hold up across different experiments, operators, or technologies. This expectation is especially important in clinical diagnostics, where technologies must undergo extensive validation and results often require orthogonal confirmation before being considered reliable. In a clinical context, you do not want to report actionable information to a physician unless you are confident in its accuracy.
This mindset provides the foundation for the project that became my recent paper. The core curriculum of my master’s program is Problem-solving and Training in Hands-On Genomics Data Science (PaTHOGEN-DS), a course designed to teach us how to develop our own experiments from initial conception to final data analysis. As part of this training, we have the opportunity to perform hands-on sequencing of bacterial genomes using both Illumina Miniseq and Oxford Nanopore Technologies (ONT) platforms, including both the GridION and MinION models. One strength of the course is that our instructors encourage us to look beyond the immediate goals of the class and explore new scientific questions within our datasets.
Motivated by this encouragement, I decided to explore methylation calling and analysis using the genomes we sequenced from two strains of Streptococcus dysgalactiae subsp. equisimilis, both isolated from patients at MD Anderson Cancer Center. Because this work began as a course project, we had a somewhat unusual dataset: 12 replicates of one strain, and 3 replicates of a second strain, all processed by six different individuals. As expected with first-time student-generated data, sample concentrations and DNA quality varied significantly. However, this variation created a natural reproducibility framework in which we could examine the effects of operator differences, DNA integrity, and sequencing platform performance.
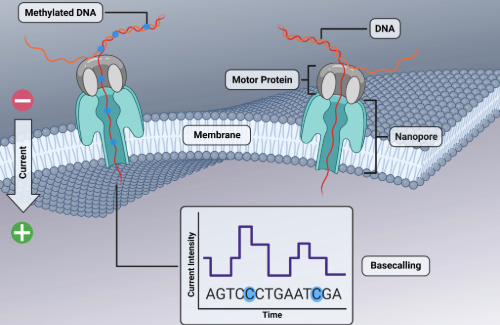
One of the major advantages of ONT sequencing is its ability to directly detect modified bases in native DNA using methylation-aware basecalling models in the Dorado software. In contrast, Illumina sequencing platforms, while widely regarded as the gold standard for sequence accuracy and assembly, cannot directly detect methylation. This contrast presented an opportunity to compare ONT-only methylome assemblies with hybrid assemblies generated from both ONT and Illumina reads. My goal was to determine how accurate, consistent, and reproducible ONT-based methylation calling truly is when benchmarked against hybrid assemblies.
Our results were encouraging. We found that ONT methylation calling and methylome assembly are highly accurate and reproducible, even across six different operators and a wide range of DNA integrity. The ONT-only methylomes performed remarkably well when compared to the hybrid assemblies. Additionally, our analysis revealed strain specific methylation patterns, suggesting potential applications in bacterial outbreak tracking and epidemiological investigations.
Learning how to perform these analyses was both challenging and rewarding. This project was my first experience developing a research study on my own, and it provided a valuable opportunity to deepen my understanding of experimental design, troubleshooting, and computational analysis. It also reinforced how much insight can be gained simply by asking, “What else can I learn from this dataset?” and following that curiosity into new analytical territory.
ONT sequencing offers rapid turnaround times, from library preparation to data analysis, which positions it as an attractive tool for future clinical and public health applications. My hope is that this work contributes to the growing body of research supporting ONT platforms and encourages continued development of high-accuracy methylation analysis workflows. Looking ahead, it is exciting to imagine a future where real-time simultaneous genome and methylome profiling may benefit public health or inform diagnostic decisions.