Accelerated clinical and technology trials in response to the Ebola virus epidemic
Issue: Halting Epidemics
07 February 2017 article
In April 2015, Josh Quick boarded an Air France flight to Conakry in Guinea. He took with him some very special luggage: all the equipment and reagents required to establish a genome sequencing laboratory in the field. Our idea was that if we could sequence Ebola virus genomes from newly diagnosed cases quickly enough then this information could provide vital information to guide the response to this tragic epidemic, which had already claimed over 10,000 lives.
Genome sequences of rapidly evolving viruses like Ebola turn out to be a rich source of information. Because the process of viral copying is error-prone, mutations in the genome are frequently introduced. We observed that Ebola, which has a 19,000-nucleotide long genome, was evolving at a rate of approximately two mutations each month. When compared with other genomes from the epidemic, clusters or lineages emerge. The clusters or lineages can highlight key parts of the DNA to help identify the groups of virus circulating in the population.
Josh, working with Miles Carroll from Public Health England, set up sequencing rapidly and sequence data started flowing back to the University of Birmingham for analysis. To do this work we used a new handheld genome sequencer from UK-based Oxford Nanopore Technologies. The sequencer, MinION, is a pocket-sized sequencer, and is powered off a regular laptop. Remarkably, this sequencer can read individual single molecules by monitoring the changes in electrical current produced when DNA blocks a tiny protein pore: ‘nanopore’ sequencing.
With just a handful of genomes we could already tell that at least two major lineages of Ebola were circulating in Guinea at the time: one branched off early in the outbreak and was the dominant type seen in Sierra Leone and Liberia. The other seemed to be mainly confined to Guinea.
A snapshot of the Ebola page from the 'nextstrain' website.
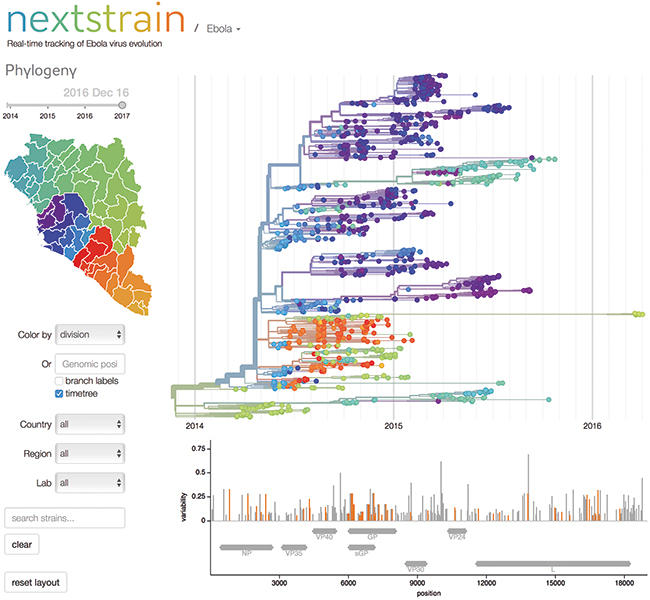
Even this broad-brush information has value for epidemiologists; the evolutionary path of viruses takes a mainly random trajectory. Two viruses that have very similar genomes must have shared a recent common host. For an epidemiologist this can be translated into simple information: patients infected with the same lineage may be in the same chain of transmission. And, crucially, a newly diagnosed patient who has a lineage 2 virus could not have been infected by a patient-carrying lineage 1.
We presented our work to the World Health Organization and the Guinean epidemic coordinators in the Ministry of Health. They were thrilled with the idea of having this information produced in real-time and invited us to continue sequencing cases until the epidemic was over. This work was done under the auspices of the European Mobile Laboratories, an EU-funded network of mobile laboratories that had been rapidly set up in Guinea and Sierra Leone to provide diagnostic support.
Next, we set up the first of our ‘genome centres’ – in Coyah, Guinea (two more were to follow, in Nongo and in Guéckédou). Sophie Duraffour, a virologist from the Bernhard-Nocht Institut, ran the sequencing facility, and rapidly trained local clinical scientists and visiting volunteers from Europe.
The teams of epidemiologists in Guinea enthusiastically received the results we generated, often within a few days of receiving a patient sample. A challenge we faced was explaining the results of phylogenetic analysis to epidemiologists not used to dealing with this type of information.
Many results were purely confirmatory, providing comfort that cases fell within known chains of transmission that were being tackled. Our data occasionally gave results that were unexpected. When combining our results with those generated by Ian Goodfellow – a virologist operating in Sierra Leone – we could show frequent examples of transmissions across the Guinean and Sierra Leone border. At the time this was not believed to be a significant factor, and resulted in tightening of border controls.
Towards the end of the epidemic, the sequencing data had their greatest value. As the gaps between new cases widened, new cases often did not have an obvious source. Often communities were unwilling to provide information about contacts. The phylogenetic information was often able to pinpoint the most likely source case directly.
When the new reported cases of Ebola in Guinea seemed to stop in October 2015 everyone breathed a sigh of relief. Yet, six months later, new cases were reported. These cases were in Forested Guinea, close to the original epicentre. It was important to understand the source of these new cases: was it a new introduction from an animal population, perhaps bats? Or had Ebola been circulating undetected in humans in that region for the past six months?
In fact, we were rapidly able to prove a third option: the case was genetically highly related to a case sequenced back in 2014. This individual had survived. The epidemiologists identified this same person as the likely source. On testing, he was positive for Ebola in his seminal fluid. Survivors with long-term Ebola infection had been known about before, including the famous case of Pauline Cafferkey who had several re-admissions to hospital with Ebola complications. Never though had there been a report of Ebola persistence for this long – 500 days – and resulting in a new cluster of cases.
The genomics and epidemiology in this case worked in perfect harmony – with only one source of information there would have been scope to doubt this result. Awareness that Ebola can persist this long and become infectious can improve the health information given to survivors. Biologically, the mechanism for this long-term persistence is not well understood and is a fruitful line of enquiry for future research.
The Ebola epidemic in West Africa from 2013 to 2016 was a remarkable event, but we now live in a time when remarkable events seem commonplace. Combinations of factors including climate change, disruption to the habitats of animal populations and increased human mobility mean that we can expect more outbreaks of emerging infectious diseases.
It took over a year from the first case to establish real-time genome sequencing in Ebola. Significant logistical challenges were encountered, including delays obtaining ethical clearance and budgets.
But genome sequencing – done rapidly and early enough – can provide vital information on where an outbreak has come from. We must improve our surveillance of emerging infectious diseases that might cause the next pandemic or epidemic so we are not caught out. In 2016 we are playing catch-up with Zika, which escaped our notice for many years until it exploded in the Americas.
Ebola survives undetected in animal populations and now in humans. We showed during the Ebola outbreak that routine sequencing was possible, and these tools should now be made routinely available for all pathogens anywhere in the world.
Nicholas Loman
School of Biosciences, University of Birmingham, Edgbaston, Birmingham B15 2TT
[email protected]