Molecular virology of SARS-CoV-2
Issue: SARS-CoV-2 and COVID-19
12 October 2021 article
Background/description of virus and family
In December 2019, cases of pneumonia were reported in patients in Wuhan, China. These patients were infected with a novel coronavirus of the Betacoronavirus lineage, ultimately designated SARS-CoV-2 due to similarities between this virus and the previously identified SARS-CoV. It seems unnecessary to reiterate the impact of this disease; however, at the time of writing, the number of confirmed infections exceeds 218 million, and 4.5 million deaths have been linked to COVID-19, the disease caused by infection with SARS-CoV-2.
Betacoronaviruses are enveloped, positive-sense single-strand RNA viruses, with genomes among the largest for RNA viruses, between 27 kb and 32 kb. Upon infection of a cell, the genome acts as a messenger RNA and initiates translation, polyproteins are produced and two virally encoded proteases process these into the mature protein forms. This processing generates a series of non-structural proteins which coordinate viral replication. One of the more interesting features of coronaviruses is the presence of a 3-to-5 exonuclease (ExoN) proofreading enzyme. This ExoN allows the viruses to reduce transcription error often associated with RNA-dependent RNA polymerases. This has allowed coronaviruses to maintain larger genomes than other RNA viruses without the risk of catastrophic mutation accumulation. Studies have demonstrated that mutations that knock out the ExoN greatly reduce coronavirus transcription fidelity and viral fitness.
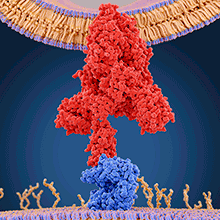
Toward the 3 end of the genome, coronaviruses encode the structural proteins, comprising the infamous spike protein (S) along with the membrane (M), envelope (E) and nucleocapsid (N) proteins. S proteins are fusion glycoproteins (gp) composed of two distinct parts, S1 and S2. The S1 domain contains the receptor-binding domain (RBD), responsible for engaging the host cell receptor, initiating the conformational changes required for fusion. The S2 domain contains a fusion peptide, which mediates the fusion between viral and cell membranes, allowing the virus to insert its RNA into the host cell.
Before SARS-CoV-2 was identified, betacoronaviruses included the clinically relevant OC43-CoV and HKU1-CoV among others. Both circulate within the human population and are among the ‘common cold’ viruses. Betacoronviruses also include SARS-CoV and MERS-CoV, diseases of significant public health concern.
What are the traits that make SARS-CoV-2 worse than other coronaviruses?
SARS-CoV-2 has characteristics typical of betacoronaviruses, though the current pandemic highlights the importance of identifying and understanding the traits unique to this virus. The S protein is essential for infection and is the dominant target for antibody responses. Given the functional role of the spike protein in initiating infection, it is possible that the key to the increased transmissibility of SARS-CoV-2 may lie within this region. Both structural and biochemical studies determined that the RBD of SARS-CoV-2 binds to the angiotensin-converting enzyme 2 (ACE2) receptor on human cells. Evidence from the first SARS-CoV outbreak identified six residues critical for effective ACE2-binding. Sequencing of the corresponding region in SARS-CoV-2 showed that five of the six residues were different from those detected in SARS-CoV. While studies are ongoing, it is likely that these changes contribute to the increased affinity of SARS-CoV-2 to ACE2 compared with SARS-CoV.
Cryogenic electron microscopy (Cryo-EM) of SARS-CoV-2 S has captured the flexibility of the S1 domain. The RBD adopts distinct conformational states, with a ‘down’ state in which the RBD is shielded from receptor binding and an ‘up’ state in which the RBD is accessible for ACE2-interaction. Interestingly, while it may be assumed SARS-CoV-2 would be more likely to have its RBD primed for receptor engagement, it appears the RBD of SARS-CoV-2 is more likely to be in the ‘down’ state. This ‘down’ state may increase the ability of the virus to evade the host immune system and, coupled with variation in the RBD residues, may have allowed the virus to balance immune evasion with infectivity.
Upon receptor binding, conformational changes in the S are required to bring the S2 region into contact with the cell membrane and initiate fusion. Coronaviruses rely on host proteases to cleave the S protein, a prerequisite for S2-mediated cell fusion. A much-discussed feature of SARS-CoV-2 is the furin-like cleavage site. While not present in other identified SARS-like coronaviruses, related betacoronaviruses (e.g. OC43 and MERS-CoV) do contain a similar furin-like cleavage domain. Given the growing number of SARS-like coronaviruses that have been isolated from animals, it is likely that more SARS-like viruses with furin-like cleavage sites will be identified in the future.
The rise of the mutants
While the ExoN activity of SARS-CoV-2 limits mutation frequency by excising nucleotide mismatches, its activity is imperfect; estimates are that functional ExoN activity can ‘repair’ 94% of mismatches. Given that an infected individual may carry 1011 virions, heterogeneity among circulating viruses is not surprising.
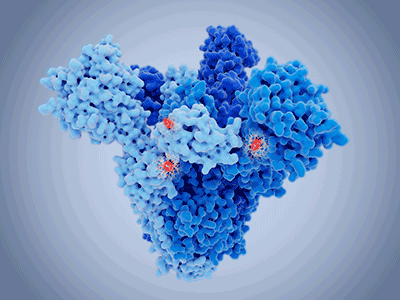
Nonetheless, specific mutations have become fixed within the population, giving rise to the variants we are familiar with. With mutations occurring throughout the genome, those localised to the S region impart clear advantages to the virus. Successful control of SARS-CoV-2 may well depend on our ability to map changes in the S protein as the primary target of vaccines.
The original SARS-CoV-2 sequence demonstrates a high affinity for the human ACE2 receptor, though computational models show that the RBD sequence was not optimal. These studies determined that asparagine was not the ideal residue at position 501 for human ACE2-binding. The N501Y substitution was among the first to be identified in what is now characteristic of the Alpha strain. This substitution was one of several changes which allowed infection rates to surge with ongoing adaptation to the human host.
Another early substitution, D614G, lies outside of the RBD, thus the mechanism for increased fitness of this variant was initially unclear. Since then, several studies have shown an increase in entry efficiency associated with the D614G mutation, with structural studies showing more of the RBD in the ‘up’ (receptor-binding) state. This mutation may represent a trade-off, wherein the virus sacrifices immune evasion for increased receptor binding.
As the pandemic progresses, virologists and public health professionals are concerned that we are now entering a phase where delays in vaccination allow the virus to continue to circulate and evolve. This new paradigm may drive selection of immune evasion mutations. An example may be the Delta variant, associated with decreased vaccine efficacy compared with the previously dominant SARS-CoV-2 strain and the Alpha variant. However, it is worth noting that all vaccines remain >60% effective at preventing infection with the Delta strain and have >90% efficacy against disease leading to hospitalisation.
The Delta variant genome has several mutations, four of which are in the S region. This includes D614G, providing increased infectivity, along with T478K and L452R. The latter two reside within the RBD, altering binding to ACE2 and providing immune evasion. These changes may represent an adaptation to the more challenging immunological landscape. The other characteristic Delta mutation is P681R, located within the furin cleavage site of the S protein. The role of this substitution is unclear, though preliminary data suggest an improved fusion efficiency and an immune evasion phenotype.
Individually, these mutations are not unique to the Delta variant; however, their simultaneous occurrence has made the Delta variant distinctive. The presence and variability of immune evasion mutations is a reminder that SARS-CoV-2 remains a significant threat; if infection numbers remain high, the virus will evolve and spread.
Further reading
Andersen KG, Rambaut A, Lipkin WI, Holmes EC, Garry RF. The proximal origin of SARS-CoV-2. Nat Med 2020;26:450–452.
Centers for Disease Control and Prevention. SARS-CoV-2 variant classifications and definitions; 2021. https://www.cdc.gov/coronavirus/2019-ncov/variants/variant-info.html [accessed 2 September 2021].
Gov.UK. COVID-19 (SARS-CoV-2) variant; 2020. https://www.gov.uk/government/collections/new-sars-cov-2-variant [accessed 2 September 2021].
Rossi GA, Sacco O, Mancino E, Cristiani L, Midulla F. Differences and similarities between SARS-CoV and SARS-CoV-2: spike receptor-binding domain recognition and host cell infection with support of cellular serine proteases. Infection 2020;48:665–669.
Shang J, Wan Y, Luo C, Ye G, Geng Q et al. Cell entry mechanisms of SARS-CoV-2. Proc Natl Acad Sci U S A 2020;117:11727–11734.
Wrapp D, Wang N, Corbett KS, Goldsmith JA, Hsieh CL et al. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020;367:1260–1263.

Keith Grehan
Faculty of Biological Sciences, University of Leeds, Leeds LS2 9JT, UK
Keith currently works at the University of Leeds as part of a team developing a virus-like particle (VLP)-based polio vaccine produced in yeast for use as a post-eradication vaccine. His other work focuses on the use of HBc as an antigen presentation system for more effective vaccine development.
What inspired you to become a microbiologist?
I started my academic life with no intentions of becoming a microbiologist. In 2012, I began an MSc studying the genetics of the innate immune system of bats. Over the course of this project, I became more and more interested in the viruses that seemed benign in bats but devastating for humans. I was struck by the impact some of these diseases have and, in some cases, how little can be done to prevent them. I was fortunate to find a PhD seeking to examine the relationship between bats and the viruses that infect them. This was my first step into microbiology, but I was hooked. The idea that understanding of viruses would lead to better treatments and prevention of disease has continued to motivate me since I first began to wonder about all these viruses that seemed to exist so well with bats.
What do you love most about your job?
This is tricky. I love research and the sense of being the first person to know something, a feeling with which most scientists are probably familiar. I also love the novelty that comes from constantly learning new things; while always challenging, the diversity of my job is one of its greatest charms. Whenever possible, I try to balance my research with teaching and science outreach. Several of my proudest accomplishments in academia have been seeing people whose training I have had a role in achieving their goals; these goals can be describing their work in a group meeting or successfully completing a PhD. Playing a role in helping someone accomplish their goals is a rare privilege that most careers don’t provide. But I think what I love most about my job is the idea that research I am involved in can be used to help people. Microbiologists at every level can be justifiably proud that their work forms part of a field that directly helps improve the lives of people all over the world.
Image: Juan Gaertner/Science Photo Library. Molecular model of a coronavirus spike (S) protein (red) bound to an angiotensin-converting enzyme 2 (ACE2) receptor (blue) on a human cell..