Only two domains, not three: changing views on the tree of life
Issue: What is life?
10 May 2016 article
In 1857, Charles Darwin sent a letter to Thomas Huxley in which he wrote: “The time will come I believe, though I shall not live to see it, when we shall have very fairly true genealogical trees of each great kingdom of nature.”
A tree for all of life – the three-domains tree
Genealogical or evolutionary trees show the relationships between organisms based upon common ancestry, like the family trees that we use to investigate our own parentage. For many biologists, Darwin’s dream was realised on the grandest scale when, in 1990, Carl Woese and colleagues proposed that all cellular life could be placed into one of three separate fundamental groups or ‘domains’ – the Bacteria, the Archaea and the Eukarya, based upon sequence comparisons of small subunit (SSU) ribosomal (r) RNA sequences. According to the ‘three-domains tree’, the Eukarya and Archaea are more closely related to each other than they are to the Bacteria (Fig. 1). Hence, in this tree our closest cousins are the Archaea, a group of micro-organisms once thought to be restricted to anaerobic and other hostile habitats like hot springs and thermal vents in the deep ocean. However, although the three-domains tree of life has dominated debate about how to organise life’s diversity at the highest level for the past 20 years or so, there is now increasing evidence that it is not the best-supported hypothesis for the evolutionary relationship between eukaryotes and Archaea.
Data and evolutionary models – how are trees made?
Although morphology has long been used to classify animals and plants, Archaea and Bacteria – which between them comprise much of Earth’s genetic and biochemical diversity – lack the wealth of morphological characters needed to reconstruct their relationships to each other or to eukaryotes. In 1965, double Nobel prize winner Linus Pauling and his collaborator Emile Zuckerkandl proposed that the sequences of the DNA, RNA and proteins found in all cells were “documents of evolutionary history” and hence were the best source of data for making global evolutionary trees. The basic procedure is to collect sequences from different species and to use a mathematical model of how we think sequences evolve to infer the evolutionary relationships between them, typically expressed in a tree diagram. Because of their central importance in the process of making trees, it is important to appreciate that all of these mathematical models use simplifying assumptions to make the analyses computationally tractable, and that they are not accurate representations of how sequences really evolve: in the words of statistician George Box, “all models are wrong, but some are useful.” In the early days of computational molecular evolution, the models used were very simple because the computers of the time were so slow. It was during this period that the three-domains tree first came to prominence, so it is interesting to ask how the tree has fared as both computers and models have improved.
Most of the models traditionally used to make the three-domains tree have assumed that the same sequences in all organisms evolve in much the same way, but this is not supported by real sequence data. For example, the nucleotide composition of SSU rRNA sequences, which are by far the most widely used molecules for making broad-scale evolutionary trees, varies dramatically in different species. This provides strong evidence that the ways in which SSU rRNA sequences have evolved in different species have changed over time. In tree building, using a model of sequence evolution that does not fit the data being analysed can often produce an incorrect tree with strong support. Recent work now suggests that this can explain why past analyses have recovered the ‘three-domains’ tree.
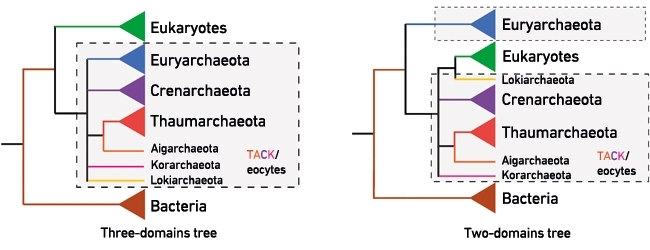
FIG. 1. THE THREE-DOMAINS AND TWO-DOMAINS TREES – COMPETING HYPOTHESES FOR THE ORIGIN OF EUKARYOTES. THE ICONIC THREE-DOMAINS TREE APPEARS IN MOST TEXTBOOKS AND DIVIDES CELLULAR LIFE INTO THREE SEPARATE MAJOR GROUPS OR 'DOMAINS': THE BACTERIA, THE ARCHAEA AND THE EUKARYOTES. IN THIS TREE THE EUKARYOTES ARE HELD TO HAVE ORIGINATED FROM A COMMON PROKARYOTIC ANCESTOR SHARED WITH THE ARCHAEA (ENCLOSED IN THE SHADED BOX). BY CONTRAST, THE TWO-DOMAINS/EOCYTE TREE RECOVERS EUKARYOTES NESTED INSIDE THE ARCHAEA WITH THE NEWLY DISCOVERED LOKIARCHAEOTA CURRENTLY THOUGHT TO BE THE CLOSEST ARCHAEAL RELATIVES OF THE EUKARYOTES. IN THE TWO-DOMAINS/EOCYTE TREE THE EUKARYOTIC LINEAGE HAD AN ANCESTOR THAT WAS ALREADY AN ARCHAEA. STUDYING UNCULTURED ARCHAEAL DIVERSITY IN NATURE THUS HOLDS THE PROMISE OF FINDING EVER-CLOSER RELATIVES OF EUKARYOTES. THE GENOMIC AND CELLULAR FEATURES OF THESE LINEAGES COULD POTENTIALLY ILLUMINATE IMPORTANT STAGES IN THE EVOLUTION OF EUKARYOTIC CELLS LIKE OUR OWN. THE THAUMARCHAEOTA, AIGARCHAEOTA, CRENARCHAEOTA AND KORARCHAEOTA ARE COMMONLY CALLED THE TACK ARCHAEA IN THE LITERATURE.
Two domains is better supported than three when new methods are used
Over the past few years, a number of new models have been developed by statisticians to try and better accommodate aspects of real molecular sequence evolution. For example, models are now available that recognise that the same sequences in different species can evolve differently in terms of their amino acid or nucleotide compositions, and other models have been developed that allow individual sites in molecular sequences to evolve in different ways to each other. Although it is widely recognised that even the best currently available models have important limitations, they fit real sequence data much better than the simpler models used in the past. Interestingly, when the new models were first used to analyse the molecular sequence data commonly taken to support the three-domains tree, an alternative hypothesis for the relationship between Archaea and eukaryotes called the ‘eocyte tree’ was better supported. In the long-neglected eocyte tree, which was first proposed by James Lake and colleagues in 1984 based upon ribosome structure, the Bacteria and Archaea can still be considered distinct primary domains but the eukaryotes originate from within the domain Archaea (Fig. 1). In other words, in the ‘two-domains/eocyte tree’, the eukaryotic lineage has an archaeal parent.
Adding new groups of Archaea increases confidence in the new tree
Microbiologists have long suspected that the micro-organisms that have been studied in the laboratory are only a tiny fraction of natural microbial diversity. The original eocyte Archaea included species like Sulfolobus (later called the Crenarchaeota in 1990 by Woese and colleagues) that live in hot acidic springs, so they were seen as rather unusual and exotic micro-organisms. In the past few years, sampling of the natural microbial world has greatly increased, driven by the availability of new molecular methods to investigate uncultured microbial diversity (Fig. 2). Recently discovered Archaea related to the eocytes include a variety of new lineages that have been informally grouped together as the ‘TACK’ Archaea. Some of the TACK Archaea have major roles in the soil and marine nitrogen cycle, suggesting that their discovery and further study is not just important because of their potential relationship to eukaryotes, but also for understanding globally important nutrient cycles. Improved sampling of lineages often has a positive impact on the accuracy of tree reconstruction, particularly if the new sequences populate parts of the tree that were previously poorly sampled. Importantly, all of the recent analyses that have included a broad sample of the new TACK Archaea have supported the two-domains/eocyte tree (Fig. 1).
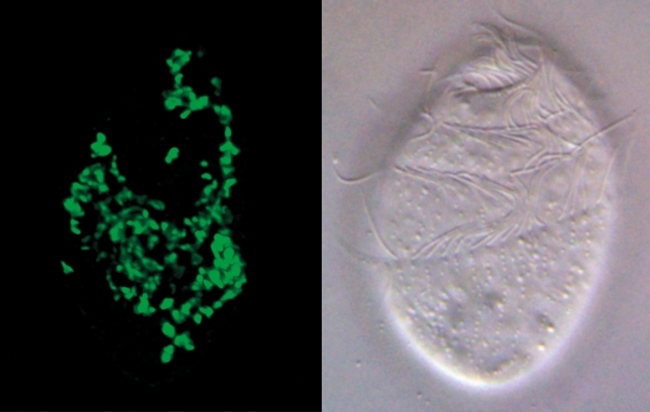
FIG. 2. MOLECULAR METHODS CAN BE USED TO IDENTIFY ENVIRONMENTAL MICRO-ORGANISMS WITHOUT CULTIVATION. THE FIGURE ON THE RIGHT SHOWS A LIGHT MICROGRAPH OF AN ANAEROBIC CILIATE PROTOZOAN CALLED TRIMYEMA THAT IS COMMONLY FOUND IN FRESHWATER PONDS IN THE UK AND ELSEWHERE. TRIMYEMA IS THE HOST FOR A PARTICULAR TYPE OF ARCHAEA CALLED A METHANOGEN BECAUSE IT MAKES METHANE. LIKE MANY ENVIRONMENTAL ARCHAEA THE INTRACELLULAR METHANOGENS HAVE NOT YET BEEN ISOLATED INTO LABORATORY CULTURE BUT THEY CAN NEVERTHELESS BE IDENTIFIED AS A SINGLE NEW SPECIES OF METHANOCORPUSCULUM BASED UPON THEIR SSU RRNA SEQUENCES, WHICH CAN BE ISOLATED AND READ USING MODERN DNA TECHNOLOGY. ON THE LEFT A FLUORESCENT DNA PROBE (GREEN) WAS USED TO CONFIRM THAT ALL OF THE MANY METHANOGENS LIVING SIDE TRIMYEMA HAVE THE SAME SSU RRNA SEQUENCE. SIMILAR PROBES CAN FACILITATE ISOLATION EXPERIMENTS BECAUSE THEY CAN BE USED TO IDENTIFY SAMPLES ENRICHED IN THE TARGET SPECIES AND ALSO TO CONFIRM WHEN A TARGET SPECIES HAS BEEN SUCCESSFULLY CULTURED. BARS, 10 ΜM.
Can the new tree help us to better understand eukaryotic origins?
The perspective on eukaryotic evolution provided by the two-domains/eocyte tree of life has already had a profound influence on ideas about how eukaryotes first evolved from their prokaryotic ancestors. Eukaryotic cells have an internal structural complexity that is not found in prokaryotes and the origins of this complexity have long been a major evolutionary puzzle. A key prediction of the two-domains/eocyte tree is that Archaea can be discovered that are more closely related to eukaryotes than the species that we already know about, and, because of this closer common ancestry, that their genomes will be more similar to eukaryotes in their protein repertoires. This prediction appears to have been vindicated by the discovery of a new archaeal lineage called the Lokiarchaeota (Fig. 3). The Lokiarchaeota are the closest archaeal relatives of eukaryotes in evolutionary trees and, consistent with that closer relationship (Fig. 1), their reconstructed genomes contain more genes for proteins that were previously thought to be eukaryote-specific. These include proteins that, in eukaryotes, are used for the cytoskeleton, in membrane remodelling and in phagocytosis, all features long-held to be unique to eukaryotic cells. At present, the evidence for the existence of Lokiarchaeota comes from metagenomes constructed from environmental DNA samples so it is now critically important to isolate viable cultures into the laboratory, to determine the cellular roles of their eukaryote-like proteins. Achieving that goal may be difficult and will require all of the classic tools of microbiology, including selective isolation, microbial physiology and cell biology, and cutting edge microscopy. However, the prize to be gained is potentially enormous because success will bring the study of eukaryotic origins much more firmly into the realm of experimental science.
FIG. 3. PART OF THE SORIA MORIA HYDROTHERMAL VENT FIELD ALONG THE ARCTIC MID-OCEAN RIDGE. THE PICTURE WAS TAKEN CLOSE TO THE LOKI'S CASTLE SAMPLING SITE FROM WHICH THE DNA SAMPLES USED TO RECOVER THE GENOME OF LOKIARCHAEOTA WERE ISOLATED. THE DETAILED METHODS USED TO RECONSTRUCT THE LOKIARCHAEOTA GENOME ARE DESCRIBED BY SPANG ET AL. (2015) NATURE 521, 173–179.

MARTIN EMBLEY
Institute for Cell and Molecular Biosciences, University of Newcastle, Newcastle-upon-Tyne, Tyne and Wear NE1 7RU, UK
[email protected]
TOM WILLIAMS
School of Earth Sciences, University of Bristol, Senate House, Tyndall Avenue, Bristol BS8 1TH, UK
[email protected]
FURTHER READING
Embley, T. M. & Williams, T. A. (2015). Evolution: steps on the road to eukaryotes. Nature 521, 169–170.
Pester, M., Schleper, C. & Wagner, M. (2011). The Thaumarchaeota: an emerging view of their phylogeny and ecophysiology. Curr Opin Microbiol 14, 300–306.
Spang, A. & others (2015). Complex archaea that bridge the gap between prokaryotes and eukaryotes. Nature 521, 173–179.
Williams, T. A., Foster, P. G., Cox, C. J. & Embley, T. M. (2013). An archaeal origin of eukaryotes supports only two primary domains of life. Nature 504, 231–236.
Image: The Human family Tree from Haeckel's Anthropogenie (1874) (French copy from 1886). Paul D. Stewart/Science Photo Library. Fig. 1. Modified from Williams et al. (2013) Nature 504, 231–236. Fig.2. Will Lewis (Newcastle University). Fig. 3. Rolf Birger Pedersen (Centre for Geobiology, University of Bergen, Norway)..