Antimicrobial Resistance Surveillance within Highly Vulnerable Patient Populations: at the Forefront of AMR Emergence
Hello, my name is William Shropshire, and I am a postdoctoral fellow working in the laboratory of Dr. Samuel Shelburne at the University of Texas MD Anderson Cancer Centre (MDACC) in Houston, TX USA. Since I joined the laboratory of Dr. Shelburne in February 2021, I have had the privilege of mentoring graduate students who do annual rotations within our lab as part of their thesis requirements through the MDACC School of Health Professions. Our latest work on the molecular epidemiology of extended-spectrum cephalosporin resistant (ESC-R) Klebsiella pneumoniae was performed by Selvalakshmi Selvaraj Anand who is now pursuing her PhD within Rice University in the Systems, Synthetic and Physical Biology (SSPB) program. Serving in my first senior author role, this was a unique opportunity for me to provide guidance as a computational biologist to a burgeoning scientist with the expectation that we perform a robust analysis of a crucial public health problem at our institution.
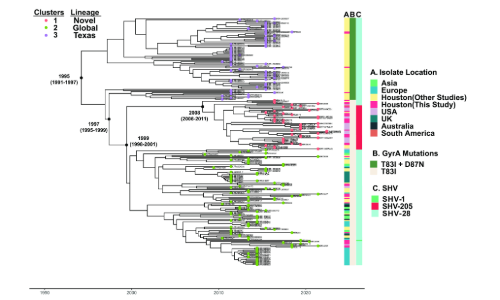
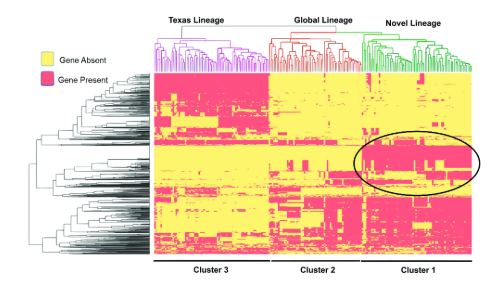
As a leading tertiary cancer centre with a global patient population, we often detect complicated infections such as those caused by ESC-R K. pneumoniae (ESC-RKp), which have large impacts on infection control. Our work and others have found that the population structure of ESC-RKp includes a highly diverse, long-branching group of heterogenous isolates that harbour commonly detected ESBL encoding genes, most notably those that belong to the blaCTX-M family. While these heterogeneous isolates typically converge towards common ESC-R mechanisms, sub-populations can have highly divergent accessory genomes, which can have wide ranging implications regarding their capacity to expand and disseminate within a hospital and community setting. One such population, which has been highly successful in disseminating worldwide is clonal group 307 (CG307) K. pneumoniae, a ‘problem clone’ that is highly associated with multi-drug resistance. Interestingly, there has been molecular clock dating analysis of CG307 that suggests this clone may have emerged within our region within the past four decades, an estimate similar to when another global problem clone, CG258, likely emerged within the United States. While previous studies of ESC-RKp in our region have noted a co-circulation of both CG307 and CG258, we were surprised to find that CG258 was rarely detected within our 5+ year study timeframe. Interestingly, we also found that the majority of CG307 isolates detected in our hospital originated from a subclade which likely diverged from the commonly detected “Texas” sub-clade isolates over two decades ago (Fig. 1). When dissecting the pan-genome, we found a highly unique accessory genome between the novel clade we detected and CG307 isolates that had been previously detected circulating in the Houston region (Fig. 2). We further found that CG307 infections caused by this novel clade occurred within patients originating from Texas as well as elsewhere in the United States, suggesting these CG307 cases were not imports from other regions of the world. There is the potential that surveillance bias in our study and others may have contributed to the stark differences in the population structure of ESC-RKp detected at our institution in comparison to other Texas Medical Center institutions. However, the fact that we detected this novel CG307 clade in recently submitted genomic data to the National Center for Biotechnology Information (NCBI) originating from the United States suggest that this subpopulation warrants further investigation.
In the past decade, we have seen a rapid shift in the affordability and feasibility of next generation sequencing for routine AMR (antimicrobial resistance) surveillance. Spearheaded through projects such as this ESC-RKp study, we are now piloting the use of Oxford Nanopore Technologies (ONT) long-read sequencing in conjunction with epidemiological data for outbreak detection. Furthermore, insights from these genomic data are being further utilised to better understand antimicrobial survival mechanisms that contribute to clinical outcome complications observed in our patient population. Given the highly immunocompromised state of our patient population, tertiary cancer centres such as ours should be a focus for potential AMR emergence. This particular observation has been further amplified by the now known impact of SARS-CoV-2 strains of interest emerging in similar patient populations. With increasing access and affordability, it is our responsibility as public health practitioners to democratize genomic surveillance across the world in order to prevent widespread antimicrobial resistance. Thus, it’s imperative that we continue to train future scientists from diverse backgrounds as we continue to tackle the dynamic challenge of AMR infections.
Image: iStock/Inventori