Decoding Huge Phage Diversity: A Taxonomic Classification of Enormous Viral Genomes
I’m Dr Ryan Cook, a postdoctoral researcher working in the laboratory of Dr Evelien Adriaenssens at the Quadram Institute Bioscience in the UK. This work was completed in collaboration with colleagues at University College London in the laboratory of Professor Joanne Santini. As viral ecologists, we are interested in the pivotal roles that viruses play in complex microbial communities, with a particular interest for those in the human microbiome.
When most people think of viruses, it is likely that they imagine well known pathogens that cause disease in humans such as SARS-CoV-2, HIV or Ebolavirus. However, most viruses do not cause disease in humans. The most numerous viruses on the planet are bacteriophages (or phages). Phages are viruses that specifically infect bacteria. As phages are well suited to target and kill specific bacteria, there is much interest in the use of phages to combat antibiotic-resistant bacterial infections and there is much research into this field. One of our areas of interest is the ecology of phages; what does the community of phages in the human gut look like? How do they interact with the bacteria in the human gut? And are they able to interact with the human gut itself?
To study communities of phages, we use viral metagenomics (or viromics). This approach seeks to separate the viruses (mostly phages) in a sample from all the other components. We then extract the genetic material from the viruses and sequence their genomes. The sequence data is then used to characterise the viral community of a given environment. This approach allows us to understand whole viral communities rather than sequencing a single virus at a time. Additionally, approaches that characterise individual viruses rely on that virus being isolated which itself relies on its host being isolated. Many bacteria are hard to isolate and grow in laboratory settings, and even when they can be grown, isolating their viruses may also be difficult. The use of viromics allows us to study the genomes of viruses that may be difficult to isolate.
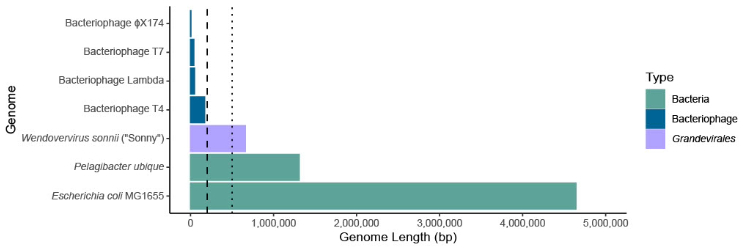
Viromics has uncovered enigmatic groups of phages in the human gut are unlike any other phages that have been isolated in the laboratory. Notably, the work of Devoto et al., (2019) uncovered the largest phage genomes to ever be reported in the human gut with lengths greater than 500,000 bases and designated them “megaphages”. Since then, megaphages have been reported in viral metagenomes derived from the faeces of humans, baboons, dogs, and horses, as well as the marine environment. To date, the largest reported phage genome is 735,000 bases in length. Perhaps the most famous megaphages are the so-called Lak phages, a group of large phage genomes that were first identified in human gut metagenomes from Laksam Upazila, Bangladesh. In addition to their large size, there is much interest in the Lak phages due to their likely use of alternate translation tables. Whilst the DNA bases TAG typically encode a stop codon (a codon that signals termination of protein translation) for most viruses and bacteria, it is likely that the Lak phages reassign the TAG codon to an amino acid such as glutamine.
We were interested in the evolutionary origins of megaphages and wanted to determine how they fit in phylogenetically with other phage genomes. Our study collected 56 phage genomes over 200,000 bases in length that were not classified to any taxonomic rank, including 23 of the so-called Lak phages. We found that the Lak phages formed a distinct grouping that contained no other phage genomes, and their nearest neighbour was another megaphage that had been identified in the marine environment named Mar_Mega_1. We examined the predicted genes for these phages and found that all 23 Lak phages likely repurpose the TAG stop codon but the marine megaphages likely do not. We proposed that the 23 Lak phages belong to their own distinct order which we have named the Grandevirales.
We further classified the proposed members of Grandevirales into two families, three sub-families and four genera. To achieve this, we performed protein clustering analyses to determine which proteins are shared amongst the phages, constructed phylogenetic trees of the shared proteins, and examined the similarity of the nucleotide (DNA) sequences of the genomes. We have since submitted our proposal to the International Committee on Taxonomy of Viruses (ICTV) who oversee the formal classification of virus taxonomy.
The classification of viruses is helpful to establish a comprehensive understanding of global viral diversity and for illustrating the evolutionary relationships of viruses. As large amounts of viral diversity have only been recovered through metagenomic and viromic approaches, we believe viruses that have not yet been cultured should still be included to inform taxonomy, such as those classified in this study. It is likely that most viral diversity remains uncharacterised, and the further study of uncultured viruses will help to shed light on the diverse viral communities in all environments.
Thumbnail image: iStock/libre de droit
References
Al-Shayeb, B., Sachdeva, R., Chen, L. X., Ward, F., Munk, P., Devoto, A., Castelle, C. J., Olm, M. R., Bouma-Gregson, K., Amano, Y., He, C., Méheust, R., Brooks, B., Thomas, A., Lavy, A., Matheus-Carnevali, P., Sun, C., Goltsman, D. S. A., Borton, M. A., . . . Banfield, J. F. (2020). Clades of huge phages from across Earth's ecosystems. Nature, 578(7795), 425-431. https://doi.org/10.1038/s41586-020-2007-4
Borges, A. L., Lou, Y. C., Sachdeva, R., Al-Shayeb, B., Penev, P. I., Jaffe, A. L., Lei, S., Santini, J. M., & Banfield, J. F. (2022). Widespread stop-codon recoding in bacteriophages may regulate translation of lytic genes. Nat Microbiol, 7(6), 918-927. https://doi.org/10.1038/s41564-022-01128-6
Cook, R., Crisci, M. A., Pye, H. V., Telatin, A., Adriaenssens, E. M., & Santini, J. M. (2024). Decoding huge phage diversity: a taxonomic classification of Lak megaphages. J Gen Virol, 105(5). https://doi.org/10.1099/jgv.0.001997
Crisci, M. A., Chen, L.-X., Devoto, A. E., Borges, A. L., Bordin, N., Sachdeva, R., Tett, A., Sharrar, A. M., Segata, N., Debenedetti, F., Bailey, M., Burt, R., Wood, R. M., Rowden, L. J., Corsini, P. M., van Winden, S., Holmes, M. A., Lei, S., Banfield, J. F., & Santini, J. M. (2021). Closely related Lak megaphages replicate in the microbiomes of diverse animals. iScience, 24(8), 102875. https://doi.org/https://doi.org/10.1016/j.isci.2021.102875
Devoto, A. E., Santini, J. M., Olm, M. R., Anantharaman, K., Munk, P., Tung, J., Archie, E. A., Turnbaugh, P. J., Seed, K. D., Blekhman, R., Aarestrup, F. M., Thomas, B. C., & Banfield, J. F. (2019). Megaphages infect Prevotella and variants are widespread in gut microbiomes. In Nature Microbiology.
Ivanova, N. N., Schwientek, P., Tripp, H. J., Rinke, C., Pati, A., Huntemann, M., Visel, A., Woyke, T., Kyrpides, N. C., & Rubin, E. M. (2014). Stop codon reassignments in the wild. Science, 344(6186), 909-913. https://doi.org/10.1126/science.1250691
Michniewski, S., Rihtman, B., Cook, R., Jones, M. A., Wilson, W. H., Scanlan, D. J., & Millard, A. (2021). A new family of “megaphages” abundant in the marine environment. In ISME Communications 2021 1:1 (Vol. 1, pp. 1-4): Nature Publishing Group.
Peters, S. L., Borges, A. L., Giannone, R. J., Morowitz, M. J., Banfield, J. F., & Hettich, R. L. (2022). Experimental validation that human microbiome phages use alternative genetic coding. Nature Communications, 13(1), 5710. https://doi.org/10.1038/s41467-022-32979-6