Demystifying Borrelia: novel computational methods to detect and identify OspA types
Lyme disease is a vector-borne infectious disease caused by the bacterium Borrelia burgdorferi sensu lato (B. burgdorferi s.l., or Burgdorferi). Infection is transferred to humans through the bites of infected Ixodes ticks. Lyme disease is estimated to affect over 600,000 people annually in the United States and Europe, with symptoms varying in severity between cases. Early indicators of Lyme disease include the characteristic erythema migrans (EM) rash at the bite site, that can occur with non-specific symptoms such as fatigue, fever and headache. Untreated early Lyme disease can progress to more serious late-stage disease that impact the skin, joints (arthritis), heart (carditis) and nervous system (neuroborreliosis). Symptoms have been shown to persist post-treatment in some individuals. Several Lyme Borrelia species are documented as pathogenic to humans, with differences in distribution between North America, Europe and Asia (Figure 1). With the exception of EM, clinical manifestations are known to differ between genospecies and consequently by geographic area. For instance, in the US and Canada, where B. burgdorferi sensu stricto predominates, Lyme arthritis is more common. In Europe, however, neuroborreliosis is more common due to the differential pathogenesis of B. garinii. In addition, borrelial lymphocytoma and acrodermatitis chronica atrophicans (ACA) occur in Europe due to prevalence of B. afzelii.

A potential Vaccine against Lyme disease
As the most common vector-borne disease in the Northern Hemisphere, there is an unmet medical need for a vaccine to protect against Lyme disease. As of this post, there are currently no approved human vaccines for Lyme disease. Pfizer and Valneva are collaborating on the development of VLA15, a six-valent candidate vaccine designed to help protect against the dominant genospecies causing Lyme disease in North America and Europe. The VLA15 vaccine candidate aims to provide protection from Lyme disease by inhibiting Borrelia Outer Surface Protein A (OspA), a protein expressed on the bacterial cell surface while in the tick vector. The investigational vaccine follows a mechanism of action based on antibodies against OspA that are introduced into the tick during ingestion of the blood meal. Binding to OspA on Borrelia in the tick midgut that aims to prevent transmission and thus infection.
History of traditional Borrelia serotyping
VLA15 is a multivalent vaccine candidate designed to provide protection against the six most common Borrelia OspA serotypes, spread across the four dominant genospecies in North America and Europe: B. burgdorferi sensu stricto (OspA serotype 1), B. afzelii (OspA serotype 2), B. garinii OspA serotypes 3, 5, 6) and B. bavariensis (OspA serotype 4). These serotypes were originally classified in the mid-1990s using a panel of monoclonal antibodies (mAbs). Unfortunately, the mAbs used to type these initial serotypes were not made widely available and new mAbs were not elaborated to classify the large number of putative remaining and emerging OspA serotypes. Over the last 20 years, an ad-hoc combination of amplicon sequencing and restriction typing of OspA has been implemented by investigators. However, while these methods can broadly differentiate serotypes, they are unable to identify sequence diversity within OspA. Such information is important invaccine design in order to assign serotypes, assess variation within serotypes, and predict immune response to new OspA variants. To this end, our team at Pfizer Vaccine Research and Development turned to next-generation sequencing (NGS) to provide higher sequence resolution of the OspA antigen. In doing so, we defined a formal typing system that is broadly available and amenable to assignment of the full range of known OspA sequences with accommodation for potential new variants.
Introducing an in silico typing method for Borrelia surveillance
Our group compiled a collection of over 400 sequenced Borrelia genomes spanning 11 genospecies that account for the majority of Lyme disease cases in North America, Europe and Asia. By leveraging phylogenetic analysis and sequence alignment, we observed clusters of OspA sequences that correlated with the known OspA serotypes. Additionally, we identified novel phylogenetic clusters outside of these canonical serotypes, many of which consist of human disease-causing genospecies. Based on this purely sequence-based classification method, we dubbed these groups OspA in silico types (ISTs). ISTs 1-8 correspond to the mAbs-determined OspA serotypes 1-8, and de-novo IST assignments (ISTs 9-17) were given to previously unclassified types, such as the human pathogenic genospecies B. mayonii and B. spielmanii (Figure 2). We further developed and released an open-source computational pipeline, the Lyme OspA in silico typing tool (github.com/pfizer-opensource/listt), to provide an automated method to determine the OspA IST of a Borrelia strain based on NGS of the ospA gene. This pipeline identifies both the in silico type and genospecies by performing sequence alignment and calculating amino acid identity to known OspA variants belonging to multiple serotypes. As this method only takes the sequence of the Borrelia ospA gene into consideration, it is highly sensitive to B. burgdorferi s.l. and avoids false positives that could be caused by other pathogens that do not carry ospA. Additionally, this means analysis can be performed in a background of host DNA, making it an ideal method for clinical surveillance as no isolation or culturing of Borrelia, a sometimes arduous task, is required.
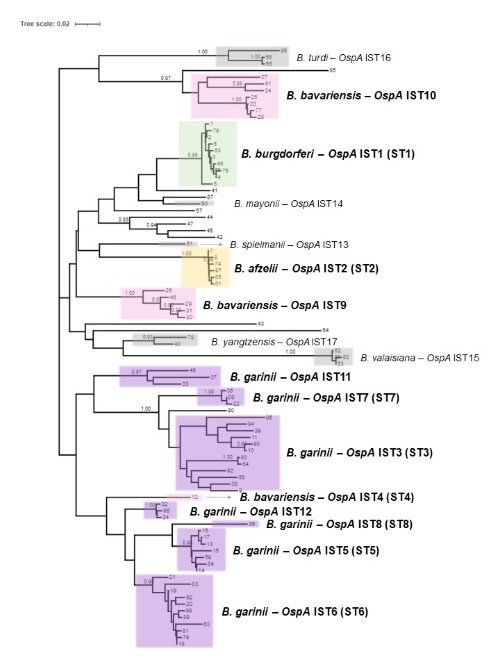
We hope that our in silico typing method will provide a valuable new tool in the Lyme disease field, both in terms of Borrelia research and vaccine design. The IST scheme has already been implemented by the public database for microbial genome diversity, PubMLST (pubmlst.org), in order to track the in silico types of novel strains as they are sequenced and deposited by contributors worldwide. As more data is collected over time, this scheme can be adapted to incorporate novel OspA variants and ISTs and provide continued monitoring of Borrelia diversity.
Image: iStock/Inventori