Papers Please: Host attribution of Salmonella Typhimurium via Machine Learning
I am Antonia Chalka, a PhD student in Professor David Gally’s group at the Roslin Institute of the University of Edinburgh, Scotland. My research applies machine learning to bacterial genome sequences in order to predict the host animal species that the bacteria have come from, known as host or source attribution. In this paper I have applied this approach to Salmonella, an important cause of foodborne illness in humans and animals. Such attribution has been a long-standing challenge in the field of microbiology as defining the likely source of an outbreak strain, chicken, cattle or pigs for example, can help control outbreaks and improve food safety.
This task is complicated by the genetic diversity within the Salmonella genus, which makes host attribution based solely on phylogeny challenging. Traditional methods have primarily relied on such phylogeny, represented through Multi-Locus Sequence Typing (MLST) or whole genome Single Nucleotide Polymorphisms (SNP) to identify the closest isolates and attribute the host based on this relationship. However, these traditional approaches have limitations, but recent advancements in machine learning have opened new doors to improve the accuracy of host attribution with genome data.
Much ado has been made about machine learning especially in the past few years, with the proliferation of algorithms and frameworks such as deep learning, and more recently, large language models like ChatGPT. Our research uses the more humble ‘Random Forest’ approach which combines multiple decision trees to make predictions. When dealing with a complex trait like host specificity, relying solely on association analysis is insufficient to paint the full picture. Machine learning allows collections of features to be considered and their relationship to each other (e.g. in the format of a decision tree), allowing us to make predictions based on a more comprehensive set of characteristics, rather than just the closest genetic relatives.
Our laboratory has been interested in studying host attribution using machine learning. Our recent paper delves into the attribution of hosts to USA Salmonella Typhimurium isolates, building upon Dr. Lupolova’s previous work. We have now employed a larger dataset and a more sophisticated approach, streamlining the process into a reusable pipeline that can be applied to similar problems in the future, such as host attribution in different pathogens or exploring bacteriophage specificity. Additionally, we aimed to identify the most effective genomic characteristics as features for building host attribution models, and we compared building models on Antimicrobial Resistance (AMR), pangenome gene clusters (PV), intergenic regions (IGR), and SNPs.
Our results were encouraging; our models were either on a par with, or outperformed traditional nearest-neighbour models. Specifically, models based on SNP data proved to be as effective as the traditional approach, whereas models trained on PV or IGR data demonstrated superior performance over phylogenetic-based assignment.
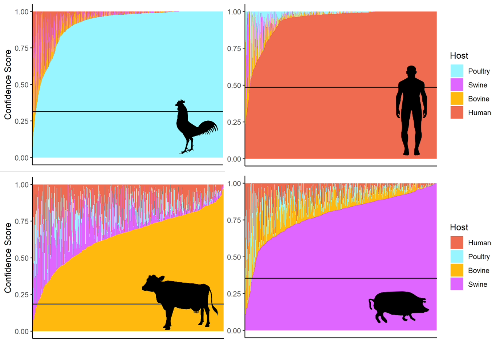
Nevertheless, our machine learning models, like traditional methods, are constrained by the population structure on which they were trained. If an isolate falls outside the clades present in the training set, the predictions may become unreliable. Though we do consider that our methodology enables a more accurate way for host assignment over traditional phylogeny-based techniques, our aim is for machine-learning-based host attribution to complement, not replace, existing approaches.
In addition to our results, we hoped to identify genes and regulatory regions responsible for host specificity by extracting them from the features deemed important during model training. This, however, proved to be a challenging and potentially misguided endeavour, given the complex web of phylogeny and genetic interactions at play.
Creating the initial models and setting up the model training pipeline presented its own set of challenges. In the world of bioinformatics, one knows that a significant part of the job is wrangling file formats and making various components work seamlessly. Automating this process was initially daunting due to the numerous tools and components that needed to come together. We used Nextflow alongside Docker to make our pipeline, which is available on Github. We used our 1.0 for our paper and are in the process of improving it with additional features and workflows.
Our long-term goal is to expand our approach and integrate it into diagnostics and outbreak investigations alongside existing techniques. Machine learning has the potential to be an invaluable tool to interrogate bacterial genomic data, and we aim to make a set of models publicly available for testing sequences. The aim is to have any interested party drop a sequence into a website and it will tell you “Oh, this probably came from a chicken (85% confidence)!” In addition, the approach can assign a human infection ‘risk’ score for any livestock Salmonella isolate but of course this score is very hard to verify – any volunteers…?
Image: David Gally/Authors