Sequencing the genome of Streptococcus pneumoniae
Posted on October 21, 2019 by Carmen Sheppard
Whole genome sequencing has revolutionised many fields of study, not least microbiology. In a short time, its use in research has significantly improved our knowledge of bacteria and viruses. Earlier this year, Carmen Sheppard hosted a workshop on whole genome sequencing of Streptococcus pneumoniae in a public health reference laboratory. This meeting was supported by funding from the Microbiology Society.

As a scientist in a microbiology reference laboratory at Public Health England (PHE), in Colindale, London, I have been involved with the transition of the national reference services for Streptococcus pneumoniae (S. pneumoniae or pneumococcus) to whole-genome sequencing (WGS) since the very beginning, starting with a pilot project to sequence just 8 genomes in 2012. Back then S. pneumoniae was chosen as a priority organism, along with Salmonella and Staphylococcus aureus for the transition of PHE’s reference services to this new technology.
S. pneumoniae is an important human pathogen, causing many disease cases worldwide despite being classed as vaccine-preventable. Vaccines against these bacteria target the protective polysaccharide (sugar) capsule that surrounds the bacterium. Unfortunately, there are more than 92 chemical variants of the capsule (known as serotypes) and vaccines can currently only provide protection against up to 23 of the most common serotypes. Accurate knowledge of serotypes that most commonly cause disease is very important to monitor how well vaccines are working and to inform decisions on future vaccines. Our lab at PHE confirms the identification and the serotype of all S. pneumoniae isolates grown in laboratories from normally sterile sites (usually blood or cerebrospinal fluid) referred to us from England and Wales. This totals over 5,000 isolates per year.
Confirming the serotype of S. pneumoniae isolates is traditionally carried out by mixing the bacteria with a large panel of antisera that are targeted against the different serotypes and observing which ones give a positive result (either by eye or under a microscope). However, this is time consuming and laborious.

Derren Ready, interim head of the PHE Central Sequencing
Laboratory gives an overview of the Service workflow.
We developed an automated bioinformatic method that analyses the WGS data from each pneumococcus and predicts its serotype from gene sequences that encode the enzymes that build the polysaccharide capsule. It took a huge amount of work to develop this bioinformatic method because some changes in these genes affect the serotype and some don’t. We published our method for serotyping by WGS (named PneumoCaT - Pneumococcal Capsular Typing) in 2016 and started using it routinely in the national PHE reference laboratory on 1 October 2017. Since then, many other laboratories have adopted this method (or derivatives of it) for their work, for example including the Global Pneumococcal Sequencing Project. One added benefit of analysing WGS data is that additional information can be easily extracted from each pneumococcus, such as confirmation that it is the correct species and has the genetic information that helps us monitor how the bacterial population is evolving over time.
Since we went live with our WGS service, we have been approached by several people from similar specialist or reference laboratories asking for advice and help with setting up WGS methods. This includes not only the technicalities of the method, but also quality assurance and validation procedures that must accompany a method that is used to issue clinical reports in an accredited reference laboratory. We decided to set up a two-day workshop in which my colleague Natalie Groves and myself – along with colleagues from our core bioinformatics department and central sequencing service – could impart our experiences and start a network of similar laboratories to help each other, discuss problems, debate solutions and best practise and to discuss data analysis methods.
The use of WGS for pneumococcal serotyping has also opened a can of worms: how should we deal with new genetic variants of serotypes? These are isolates with changes in the gene coding for the enzymes that create the polysaccharide, each of which must be checked to determine whether the genetic change affects the serological type of the organism (the polysaccharide). Scientific literature has become a hotbed of publications about genetic serotype variants, some that do, some that do not, alter the polysaccharide structure of the capsule, to the point where it is starting to get hard to know ‘When is a serotype a serotype?’. We decided that one of the main topics of the workshop could be about serotype variants, their nomenclature, and how to deal with them as a reference laboratory, their meaning for epidemiology and vaccine surveillance.

Matthew Goulden from the PHE Core Bioinformatics team gives an overview of the bioinformatic pipelines and quality considerations for routine workflows.
The workshop was advertised on Twitter and was free to delegates, though with limited spaces available. In the end, we had 16 participants from laboratories around Europe and one who travelled from the USA. We received a grant from the Microbiology Society to cover travel expenses for external speakers.

Natalie Groves from our laboratory gives an overview of methods we have assessed and used for analysing the first full year of routine S. pneumoniae sequencing data.
We invited Dr Stephanie Lo from the Wellcome Sanger Institute to talk on the newly published Global Pneumococcal Sequencing Clusters, and how to use them to contextualise local WGS data. Dr Andries Van Tonder, formerly from Sanger, was invited to talk about his work on the many serotype variants discovered in the Global Pneumococcal Sequencing project, and Dr Nick Croucher from Imperial College London was invited to give us the low down on the use of genomic population clustering methods and the use of WGS data to compare different pneumococcal populations.
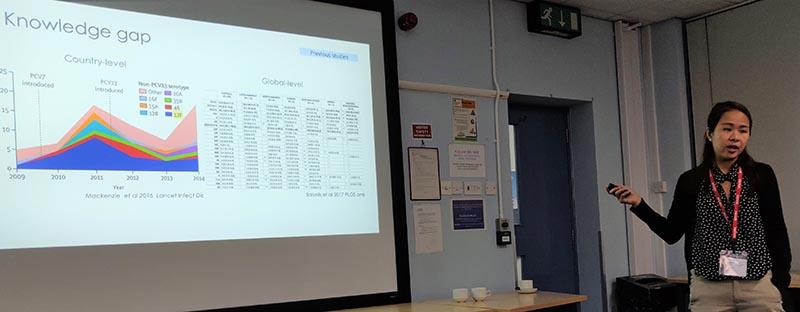
Stephanie Lo from the Wellcome Trust Sanger Centre gives an overview of the Global Pneumococcal Sequencing project and methods to compare local data with the global genome collection.
It was an exhausting but highly successful couple of days. Everyone was appreciative of the level of information and detail shared by ourselves, our colleagues, the other participants and by our external speakers.

Andries Van Tonder, formerly from Sanger gives an overview of some of the serotype variants found in the global collection.
We had plenty of interaction, questions and suggestions. We had spirited debates regarding the nomenclature of serotypes and all delegates decided that as there was a lack of an ‘overarching official body’ to decide on these, so one should be set up.

Nick Croucher from Imperial College London gives an update on analysis methods for assessing pneumococcal populations.
This could be a committee with criteria that needed to be fulfilled before a new serotype could be named and would ensure naming conventions are applied. For example, variants that were no different by serology or polysaccharide structure, but had significant differences in genetic structure, should have a different genetic variant nomenclature that should not be confused with the serologically-based type.
We were very pleased with how the workshop went and have plenty of actions to take forward.
To quote feedback from one of our participants “Thank you so much, Carmen. …., this is the most informative meeting I ever attended. A lot of sharing, discussion and debate which I found very valuable. Many thanks to you, Natalie and everyone else who are involved in hosting this event. What a fantastic meeting!” So, it looks like we will have to run it again sometime!