Can we identify potential viral zoonoses before they cross the species barrier?
Issue: Zoonotic diseases
05 November 2015 article

The toolkit for the identification of zoonotic viruses has evolved substantially over the last century. In the early days of pathogen research, the presence of a viral pathogen was suspected if filtered bodily fluids could be used to infect experimental animals (filtration was used to remove bacterial pathogens). Subsequently, serological assays, in vitro culture in cell lines, such as Vero cells, and the polymerase chain reaction (PCR) were added to the armamentarium.
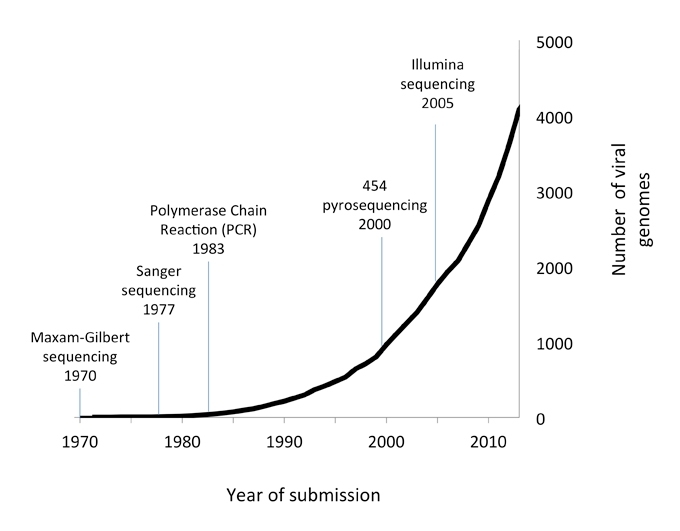
The field is now being revolutionised by next generation sequencing (NGS), an extremely powerful technique that can be used to generate millions of sequence reads in a matter of hours. As little as 1 ng of DNA can be used to identify multiple pathogens in a sample, without the need for specific primer-based amplification. Since NGS became routinely available at the start of the millennium, the number of new viral sequences deposited in the NCBI database has risen exponentially (Fig. 1).
FIG. 1. NUMBER OF VIRAL GENOMES SUBMITTED TO NCBI 1970–2014.
History of the discovery of zoonotic viruses
In the 19th century, Louis Pasteur realised that rabies was caused by an agent that could not be visualised using a light microscope. Dmitry Ivanovsky and Martinus Beijerinck later showed that fluid passing through a porcelain Chamberland filter (Fig. 2) contained an agent infectious to tobacco plants. Beijerinck called this agent a virus although it was not until the 1930s that the structure of the tobacco mosaic virus was described using electron microscopy and X-ray crystallography.
FIG. 2. CHAMBERLAND-PASTEUR FILTER.

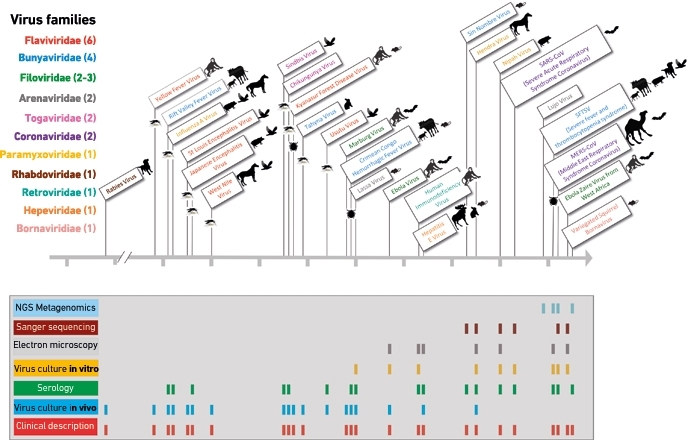
During the 20th century, inoculation of experimental animals and tissues with filtered materials from infected individuals allowed scientists to postulate that viruses were responsible for several zoonotic infections. In 1927, blood samples from humans with yellow fever were used to infect Macacus sinicus and Macacus rhesus monkeys (this was the first rhesus monkey experiment). These were further passaged in vivo by inoculation of filtered plasma or exposure to Aedes aegypti mosquitoes. The likely viral aetiology of yellow fever was solved in this series of experiments, lasting only three months. Sadly, the senior author of the paper, Adrian Stokes, was accidentally infected during the study and died of yellow fever prior to publication of the report. Shortly after this, Rift Valley fever was identified in sheep in a farm in rural Kenya. Dilutions of plasma filtered through Berkefeld and Chamberland filters were found to infect sheep, cows and a goat experimentally (but not horses or pigs), even after 54 days of storage. In this study, all the investigators also subsequently became infected and, in an experiment reflecting the ethical standards of the day, a deliberate infection of a patient suffering from recurrent malaria was carried out to see if he would be cured; he was not - he developed Rift Valley fever and then relapsed shortly afterwards with severe malaria. In both of these studies, Koch’s postulates were fulfilled for the first time for viral infection. However, sequencing of the viral genomes of these organisms was not carried out until 1985, using cloning and chemical sequencing.
In vitro techniques for culturing viruses were developed in the middle of the 20th century. Vero cells derived from Chlorocebus African green monkey kidneys became available in 1962 and Lassa fever was one of the first zoonotic viruses to be identified using Vero cell culture in 1970 by Buckley and Casals. The presence of a virus was confirmed by filtration and in vivo infection in experimental mice (rodents of the Mastomys natalensis species were later found to be the natural reservoir; Fig. 3). By this time, a number of additional techniques were available to confirm infection. Serological assays including complement fixation and neutralisation assays were carried out, and the virus was additionally visualised by electron microscopy. Two of the laboratory workers in Buckley and Casals lab at the Yale Arbovirus Research Unit (YARU) in New Haven became infected and one died of the illness.
FIG. 3. THE MULTIMAMMATE MOUSE MASTOMYS NATALENSIS.

Sequences of viral pathogen genomes were not commonly published until the 1980s. HIV infection was sequenced at the time of discovery by Francoise Barré-Sinoussi and colleagues although the sequence itself was not actually included in the first report. Later virus discovery publications have almost always described the viral genetic sequence. When related genomes have been available, a common approach has been to amplify viral genomic segments using random or conserved PCR primer sets from multiple virus families. Sin Nombre virus was identified in clinical samples and Peromyscus maniculatus deer mice after an outbreak of severe respiratory illness in patients in the USA in 1993 using conserved hantavirus PCR primers. In 1994, PCR-based sequencing using conserved primers from the Paramyxoviridae family allowed identification of Hendra and Nipah viruses. Hendra was identified during an outbreak of severe neurological and respiratory illness in horses and stable workers near Brisbane and an outbreak of encephalitis in humans and respiratory illness in pigs in Malaysia was found to be associated with the closely related but distinct Nipah virus. This use of conserved primers is based on a best guess of which primer sets to use to detect an unknown pathogen and requires that these bind to conserved regions of viral genetic material present in infected samples. The advent of unbiased next generation sequencing (sequencing of all the RNA or DNA in a sample including human- and pathogen- derived DNA and RNA) that is not based on the presence of specific PCR primers means that many more pathogens are likely to be discovered over the coming years.
Next generation sequencing
One of the first emerging zoonotic viral pathogens to be identified using unbiased NGS was Lujo virus, following an outbreak of viral haemorrhagic fever in five individuals (an index case from Zambia, a paramedic, two nurses and a cleaner). Within 72 hours, a new arenavirus was identified. This was one of the first studies to harness the power of NGS to make a rapid diagnosis during an outbreak. Unbiased Illumina NGS during the recent outbreak of Ebola virus in West Africa also resulted in the rapid publication of multiple full genome sequences, contributing hugely to the international effort to develop a vaccine and other therapeutic interventions.
Predicting the next epidemic
NGS is emerging as a powerful technique for the identification of infectious pathogens and has great potential as a surveillance tool. It has been estimated by scientists at Columbia University that as many as 320,000 viruses in mammals remain undiscovered. In a single study, 58 new viruses from nine virus families were identified in a single species (the Indian flying fox bat Pteropus giganteus). The majority of emerging viral pathogens are zoonotic and future research now aims to identify viruses in the zoonotic pool before they transit into human populations. This pool is deep, however, and much work remains to be carried out. While some investigators aim to target outbreaks of disease causing deaths in wildlife, zoonotic pathogens such as Ebola virus may not cause disease in their natural host, so predicting the possibility of spillover events before they happen will require surveillance of high-risk reservoir hosts such as bats. Such events are more likely to occur in association with changes in environmental equilibrium; deforestation, for example, may have contributed to the recent Ebola outbreak in West Africa, resulting in increased contact between bats and humans.
Several ongoing studies such as the Wellcome Trust VIZIONS project are now attempting to identify pathogens present in animal reservoirs that have the potential to transfer across the species barrier into humans. Combining high-throughput NGS data with functional virological assays, for example, whether or not a cognate receptor is present in humans, may help us to predict which viruses present a significant risk to the human population. Canine distemper virus, as an example, has crossed from dogs into several other species including seals, tigers, foxes and non-human primates but has not entered the human population despite the presence of the receptor in human cells. Many other potential pathogens await discovery.
FIG. 4. EXAMPLES OF ZOONOTIC VIRUSES AND METHOD OF DISCOVERY OVER TIME.
HANNA JEROME, SREENU B. VATTIPALLY & EMMA C. THOMSON
MRC University of Glasgow Centre for Virus Research, Sir Michael Stoker Building, University of Glasgow, 464 Bearsden Road, Glasgow G61 1QH, UK [email protected]
[email protected]
[email protected]
Acknowledgements
Emma Thomson is funded by the Wellcome Trust and the Medical Research Council (MRC).
FURTHER READING
Anthony, S. J. & others (2013). A strategy to estimate unknown viral diversity in mammals. mBio 4, e00598-13.
Briese, T. & others (2009). Genetic detection and characterization of Lujo virus, a new hemorrhagic fever-associated arenavirus from southern Africa. PLoS Pathog 5, e1000455.
Daubney, R. & others (1931). Enzootic hepatitis or rift valley fever. An undescribed virus disease of sheep cattle and man from east Africa. J Pathol Bacteriol 34, 545–579.
Gire, S. K. & others (2014). Genomic surveillance elucidates Ebola virus origin and transmission during the 2014 outbreak. Science 345, 1369–1372.
Sakai, K. & others (2013). Canine distemper virus associated with a lethal outbreak in monkeys can readily adapt to use human receptors. J Virol 87, 7170–7175.
Image: Fig. 2. Division of Medicine & Science, National Museum of American History, Smithsonian Institution. Fig. 3. Tom McHugh/Science Photo Library. Fig. 4. Hanna Jerome, Sreenu B. Vattipally & Emma C. Thomson..