A phylogenomic framework for understanding Streptomyces in agricultural settings
Understanding how pathogens evolve, spread, and maintain the molecular pathways necessary for virulence is critical for predicting the risk of disease emergence. As a research plant pathologist with the United States Department of Agriculture Agricultural Research Service, I am charged with improving our ability to manage common scab disease of potato. Common scab is an economically costly soilborne disease of potato and other tuber and root crops caused by several lineages of Streptomyces bacteria. The pathogen is endemic in many potato-growing regions, thanks, in part, to their ability to form spores that can survive in the soil in the absence of a host plant. The major challenge for developing management tools against common scab is that the disease is caused by more than ten phytopathogenic species of Streptomyces and their diversity is largely uncharacterised. A further complication is that the primary pathogenicity determinant for Streptomyces, the pathogenicity island that encodes the biosynthetic genes for the phytotoxin Thaxtomin, can transfer among strains enabling the emergence of novel pathogens.

A high-resolution phylogenomic framework is necessary to understand Streptomyces diversity, population structure, evolution, and transmission. Such knowledge is critical for enabling disease risk prediction as well as developing focused, location-specific disease management interventions. My lab at the Agricultural Research Service (ARS) houses a collection of more than 2000 plant-associated Streptomyces including more than 1000 strains that have the Thaxtomin biosynthetic genes. Generating the necessary genomic resources required a large collaborative project that relied on the substantial bacterial genomics expertise of Jeff Chang and Alexandra Weisberg (Oregon State University, USA). We sequenced 166 new Streptomyces genomes, primarily from the ARS culture collection. Though more than 1000 Streptomyces genomes are available in GenBank, only a handful are from plant-pathogenic strains before our project. Multiple novel species-level groups were present in the newly sequenced strains, indicating that we are still scratching the surface of understanding the diversity of plant-pathogenic Streptomyces.

Because the primary phenotype we were interested in is plant pathogenicity, we focused on the mobile genetic elements that are associated with disease, primarily the Thaxtomin biosynthetic genes. Through robust classification of mobile genetic elements, we identified novel types of virulence-associated mobile genetic elements, novel insertion sites harbouring mobile genetic elements in the chromosome, and novel combinations of virulence genes within strains. Moreover, we were able to determine how the elements spread among strains. Importantly, the analyses suggest that horizontal gene transfer of the Thaxtomin biosynthetic genes is relatively rare, in contrast to the current paradigm in the field. Sequencing of closely related pathogenic and non-pathogenic strains revealed the loss of the Thaxtomin mobile genetic elements and explains the polyphyletic distribution of the genes in the population more parsimoniously than rampant horizontal gene transfer.

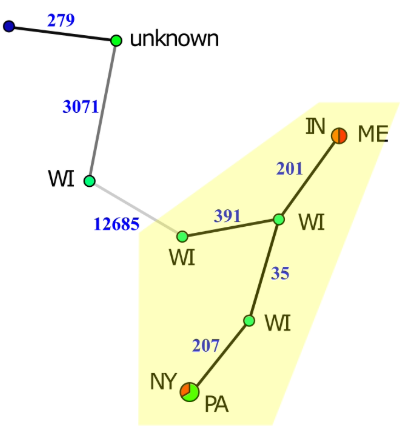
Another exciting aspect of the work was the use of genome sequences for epidemiological studies. Construction of minimum spanning networks of the whole genomes of select species of Streptomyces identified some transmission patterns of the pathogen. Within Streptomyces stelliscabiei, two instances of very recent transmission were apparent as indicated by two nearly identical strains being isolated from two distinct states (see figure, circles indicate genotypes and colours indicate state of isolation; therefore, multicolour circles indicate the same genotype isolated from two different states indicating very recent transmission of the pathogen). Overall, however, very few recent transmission events were identified. In contrast, relatively recent transmission events were common as indicated by multiple genotypes that are closely related to one another (see yellow highlighting in figure indicating different genotypes separated by 35 to 391 single nucleotide polymorphisms) being isolated from multiple U.S. states. One potential explanation for this transmission pattern is that spread of the pathogen among grower sites used to be common but has become rare. Starting in the early 1900s, North America implemented certified seed potato production to mitigate the transmission of diseases. It is possible that efforts to ship only clean seed potatoes dramatically curtailed more recent pathogen transmission.
In all, through collaborative analyses of a large collection of Streptomyces genomes, we drastically expanded our understanding of the diversity, evolution, and transmission of the pathogen and the mobile genetic elements responsible for disease manifestation. But common scab will continue to remain a devastating problem for potato growers, at least in the near future. Our results suggest that emergence of novel lineages of the pathogen through horizontal gene transfer occurs relatively rarely. But the pathogen has already diversified significantly and is endemic and many potato growing regions. Our results provide a framework for classification of the pathogen and its mobile genetic elements. But surprisingly, when we performed controlled greenhouse pathogenicity assays for 60 of our sequenced strains, we were unable to identify clear patterns to enable prediction of virulence based on mobile genetic element subtypes or species classification. Our greenhouse assays do support the paradigm in the field that the key to mitigating common scab disease symptoms is to mitigate the impact of the Thaxtomin toxin, a challenging problem. Nevertheless, we now have the tools and opportunity to characterise field populations of the pathogen, develop specific molecular diagnostic tools, improve our understanding of potato cultivar-specific disease resistance, and help mitigate its effect.
Image: Joseph Mowery (ARS)