Inside the Gut Microbiome: Investigating the Bacterial Hosts of Antibiotic Resistance Genes
My name is Dr Greg McCallum, I’m currently a postdoctoral research associate at the University of Liverpool, UK. I completed my PhD at the Institute of Microbiology and Infection, University of Birmingham, UK, in the laboratory of Professor Willem van Schaik, where I researched antibiotic resistance in the human gut microbiota.
Antibiotic resistance, where bacteria become resistant to the drugs we use to treat infection, is one of the largest global health threats facing humanity. As more bacteria become resistant to antibiotics, the number of people dying from infections is rapidly rising globally. Random DNA mutations can allow bacteria to gain resistance to antibiotics. But, more importantly, antibiotic resistant genes can be spread between bacteria on small pieces of DNA called plasmids. This process is termed horizontal gene transfer, and it occurs in many ecosystems, including the human gut microbiota.
The human gut microbiota is made up of hundreds of species of bacteria living within the human intestinal tract. Most of these bacteria have a commensal or mutualistic relationship with their host. Humans, therefore, benefit from the gut microbiota aiding digestion and shaping the immune system. However, the gut microbiota also contains opportunistic pathogens. These opportunistic pathogens are species that can live in the gut of a healthy individual without causing harm, but in some cases can take the opportunity to cause infection when the host becomes immunocompromised or otherwise vulnerable. In the last few decades, there has been a substantial rise in the number of infections caused by antibiotic resistant opportunistic pathogens originating from the gut.
As the gut microbiota lives in an environment that encourages horizontal gene transfer, it is hypothesised that opportunistic pathogens may be acquiring antibiotic resistance from commensal bacteria. Gut microbiota research often utilised metagenomic sequencing, where all DNA from a complex sample is sequenced to find what is present. This technology has revealed that the human gut microbiota contains many antibiotic resistance genes, collectively termed the human gut resistome. Metagenomic sequencing also provides information of which species of bacteria are present in the sample. However, linking those data to see which bacteria are carrying those resistance genes is not trivial, particularly if the resistance gene is present on a plasmid, separate from the bacterial chromosome.
My PhD focused on using a technique called metagenomic chromosome conformation capture (termed meta3C or Hi-C), to link resistance genes to their host to see the extent to which our commensal bacteria are carrying and transferring antibiotic resistance genes in the human gut. Meta3C/Hi-C works by physically cross-linking DNA inside cells before sequencing, allowing us to find links between antibiotic resistance genes and pieces of chromosomal DNA that can then be used to identify the species of bacteria carrying the gene.
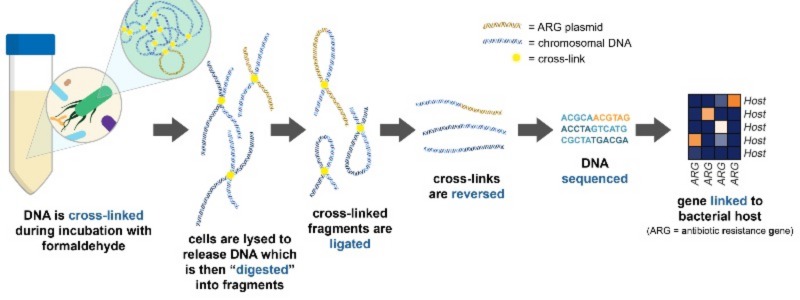
How meta3C/Hi-C works. DNA is physically linked before sequencing, allowing us to investigate the bacterial hosts of certain sequences of DNA by finding these links in the data. Adapted from Figure 1.
Our recently published work focussed on creating a workflow for analysing meta3C/Hi-C data for linking resistance genes to their bacterial hosts in the human gut. During analysis of our own meta3C data on a human faecal sample, we found artefacts in the results that could falsely link genes to an incorrect species. Whilst developing the analysis workflow, we re-analysed data from other published studies that performed meta3C/Hi-C on gut microbiota samples, and found that these artefacts were present in all meta3C/Hi-C data. In order to reduce the impact of these artefacts, we developed steps to remove noise in the data during analysis, including removal of links to repetitive regions of the genome, and requiring at least five unique links between sequences to be considered a “true” link.
Using this novel analysis workflow, we were able to link 87 antibiotic resistance genes to their bacterial hosts across the datasets. These links revealed that genes coding resistance to aminoglycoside and tetracycline antibiotics were widespread in commensal species of the human gut, indicating that commensal gut bacteria are an important reservoir for resistance genes.
Future work aims to further develop this analysis workflow to improve the classification of the linked hosts to allow more bacterial hosts of antibiotic resistance genes to be revealed. This characterisation of the human gut resistome continues to be important to finding how opportunistic pathogens are becoming resistant to antibiotics, so we can better know how to prevent the emergence of drug resistant strains that threaten future global health.
Image: Authors