Uncovering the fungal pangenome
Posted on July 4, 2018 by Charley McCarthy
This June, Charley McCarthy presented his poster ‘The pangenome of Aspergillus fumigatus’ at the Early Career Microbiologists Summer Conference in Birmingham. Charley is in the third year of his PhD at Maynooth University, supervised by Dr David Fitzpatrick. For those of you that couldn't attend the conference, here, Charley discusses his research.
I am based in the Genome Evolution Lab at Maynooth, and our research is mostly focused on the evolution of fungi and fungal-like oomycetes.
I’m primarily a bioinformatician, having caught the bug during my undergraduate thesis, and so most of my research takes place in the 'dry lab'. Despite never having much formal programming experience prior to my PhD I always had a knack for computers, and being able to use computer programming to research evolution is something of the best of both worlds for me.
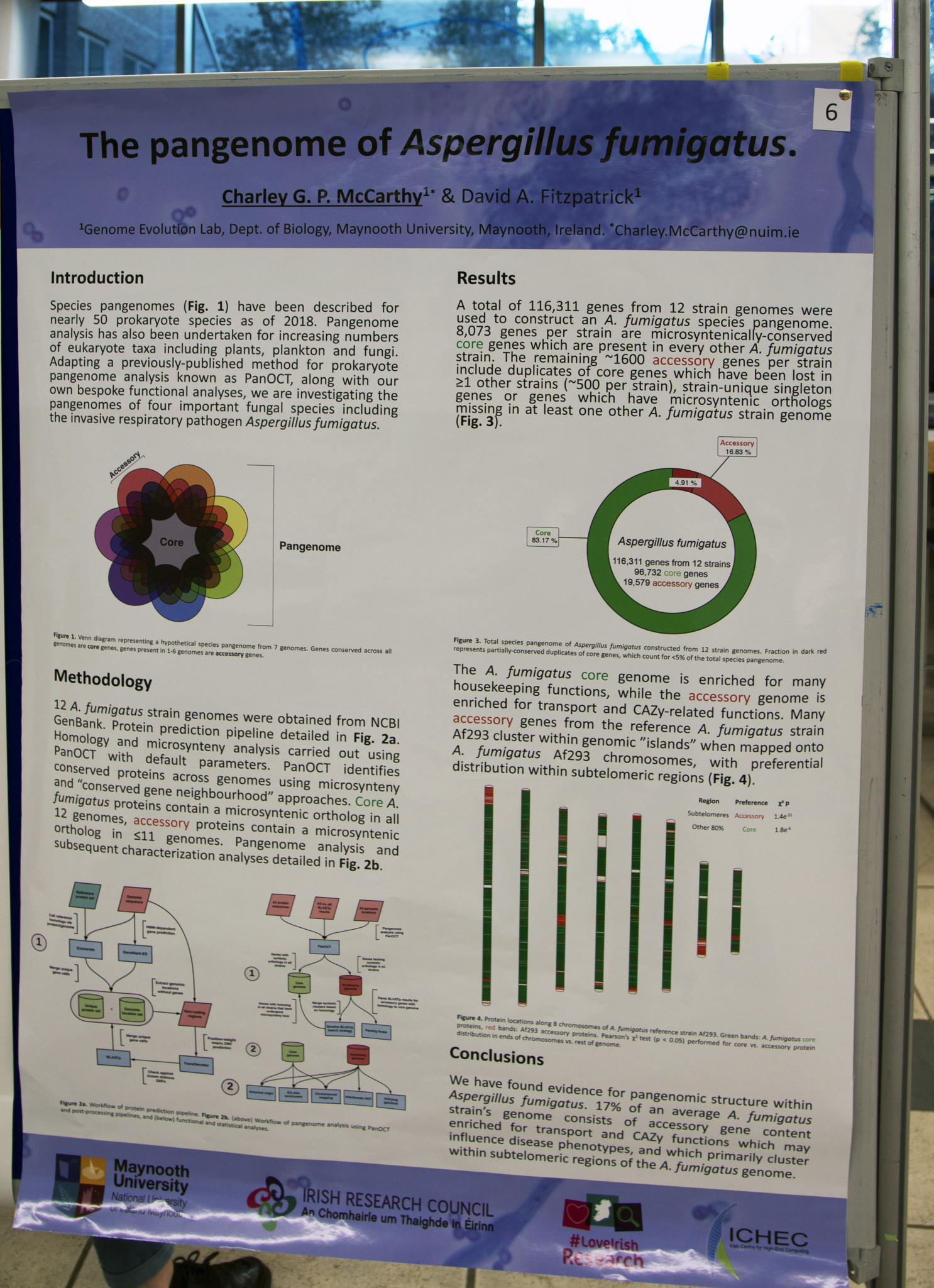
At the ECM Forum Conference, I was presenting a poster detailing some of our recent research into genomic variability in fungi. We are focused on a few important fungal species and the potential influence of this variability on their adaptation and evolution.
As genome sequencing has become more cost-efficient it has become common for some species to have multiple strains or isolates sequenced. These strains are usually from different environments and their genomes often have unique genetic information that may not be reflected in the reference genome. To account for such variation within species, the concept of a species 'pangenome' was developed in the mid-2000s.
Initially, research into species pangenomes was focused on bacteria and archaea. An increasing number of eukaryotic pangenomes have been described including those from plant, plankton and fungal species. We have been investigating the pangenomes of a number of important fungi, including Aspergillus fumigatus.
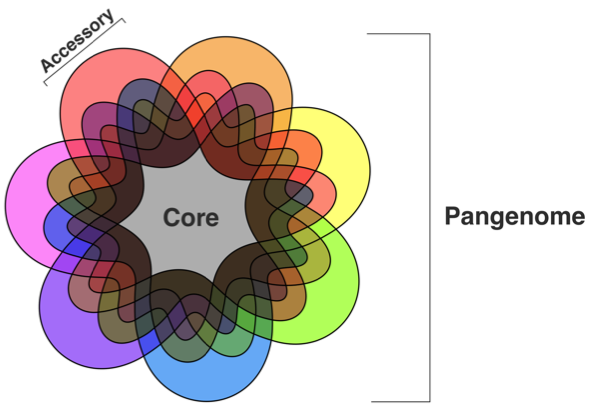
A species’ pangenome can be divided into its 'core' genome
(containing all genes conserved throughout a species),
and 'accessory' genome (containing genes variably
distributed among strains)
A. fumigatus is an opportunistic respiratory pathogen. We constructed an A. fumigatus species pangenome using whole genome data from 12 publicly-available genomes including both clinical and wild-type strains. We used computational methods to define species’ core and accessory genomes based on shared genomic location.
A given strain of A. fumigatus encodes between 9,500 to 10,000 genes. Just over 8,000 are core genes, between 80-85% of an average strain’s gene set. The remaining genes are accessory genes, which range from strain-unique singleton genes to genes missing an from only one other strain. Some of these accessory genes appear to be duplicates of core genes which have been subsequently lost in some strains but retained in others.
We’re currently characterizing the core and accessory genomes of A. fumigatus using a variety of different methods. Our results so far suggest that some processes such as small molecule metabolism (which can encompass secondary metabolism and toxin production) are conserved core processes with all strains. Meanwhile, other potential infection processes - including those involved in protein transport or antimicrobial resistance - are variably distributed. Many accessory genes cluster towards the ends of chromosomes; areas that are known to mediate the infection process of A. fumigatus in human hosts.

- All proteins encoded along chromosome 1 of A. fumigatus Af293. Red bands correspond to accessory genome clusters, green bands correspond to core genome clusters and white bands are non-coding DNA
With our research of the A. fumigatus pangenome and other fungal pangenomes, we hope to uncover the genomic origins of variation within fungal species and their consequences for strain adaptation and evolution. This may then be useful for locating regions of DNA that could be targeted to deter infection by invasive fungal pathogens like A. fumigatus. Hopefully, we will also encourage more large-scale comparative analysis of strains of eukaryotic species to match the levels of analysis for prokaryotic species.
Charley published his research in Microbial Genomics. Read his research article here.