Prions
A prion is a type of protein that can cause disease in animals and humans by triggering normally healthy proteins in the brain to fold abnormally.
The prion mode of action is very different to bacteria and viruses as they are simply proteins, devoid of any genetic material. Once a misfolded prion enters a healthy person – potentially by eating infected food – it converts correctly-folded proteins into the disease-associated form. To date, nobody knows quite how this happens.
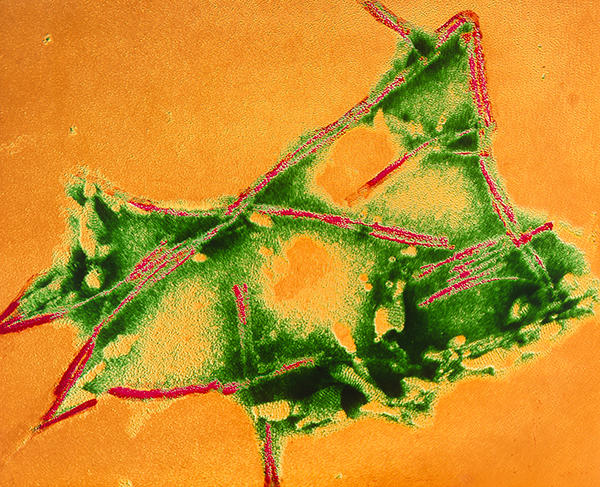
Prions in "mad cow" brain. Coloured transmission electron micrograph (TEM) of prion fibrils in the brain of a cow infected with BSE (Bovine Spongiform Encephalopathy) or "mad cow" disease. Prions are virus-like organisms made up of a prion protein. These elongated fibrils (green) are believed to be aggregations of the protein that makes up the infectious prion. Prions attack nerve cells producing neurodegenerative brain disease. "Mad cow" symptoms include glazed eyes and uncontrollable body tremor. Prions cause BSE in cattle; scrapie in sheep and goats; and Creutzfeldt-Jakob disease in humans.
Learn more about prions
-
Using X-rays to look inside the cell
Protein misfolding disorders are a class of diseases associated with unchecked protein misfolding and aggregation. The two examples we focus on are Huntington’s disease and Alzheimer’s disease, which are both neurodegenerative, protein misfolding disorders and share common features.
-

New techniques detect BSE in extra neuronal tissue of cattle
Bovine spongiform encephalopathy (BSE) or “Mad Cow Disease,” as it is more publicly known is a transmissible neurodegenerative disease affecting cattle. Importantly, it is also transmissible to humans and there is currently no treatment available. Scientists around the globe are working to better understand the infectious agent that causes the condition – a misfolded prion protein (PrPSc).
-

Prions, vCJD and the immune system relay
Despite more than a decade’s worth of research, many aspects of variant Creutzfeldt–Jakob disease (vCJD) still remain a mystery. This fatal neurodegenerative disease of the brain is often referred to as the human form of bovine spongiform encephalopathy (BSE), known to most as ‘mad cow disease’.
-

Spotlight on Grants: A search for prions in yeast
Prions are a special class of protein that can exist in two forms: normal and misfolded. Misfolded prions can act as infectious agents and have been linked to brain diseases such as human Creutzfeldt-Jakob disease and mad cow disease (bovine spongiform encephalopathy). Not all prions cause disease though, as is the case with yeasts, where several different prions have been described.